
Abstract
Abstract
Regenerative medicine is in need of solid, large animal models as a link between rodents and humans to evaluate the functionality, immunogenicity, and clinical safety of stem cell–derived cell types. The common marmoset (Callithrix jacchus) is an excellent large animal model, genetically close to humans and readily used worldwide in clinical research. Until now, only two groups showed the generation of induced pluripotent stem cells (iPSCs) from the common marmoset using integrating retroviral vectors. Therefore, we reprogrammed bone marrow–derived mesenchymal cells (MSCs) of adult marmosets in the presence of TAV, SB431542, PD0325901, and ascorbic acid via a novel, excisable lentiviral spleen focus-forming virus (SFFV)-driven quad-cistronic vector system (OCT3/4, KLF4, SOX2, C-MYC). Endogenous pluripotency markers like OCT3/4, KLF4, SOX2, C-MYC, LIN28, NANOG, and strong alkaline phosphatase signals were detected. Exogenous genes were silenced and additionally the cassette was removed with a retroviral Gag precursor system. The cell line could be cultured in absence of leukemia inhibitory factor (LIF) and basic fibroblast growth factor (bFGF) and could be successfully differentiated into embryoid bodies and teratomas with presence of all three germ layers. Directed differentiation generated neural progenitors, megakaryocytes, adipocytes, chondrocytes, and osteogenic cells. Thus, all criteria for fully reprogrammed bone marrow–MSCs of a nonhuman primate with a genetically sophisticated construct could be demonstrated. These cells will be a promising tool for future autologous transplantations.
Introduction
Embryonic stem cells (ESCs) undeniably have immense capability for use in regenerative medicine, but there are still ethical concerns. In contrast, the recently produced induced pluripotent stem cells (iPSCs) could be an alternative source for such cell therapies (Takahashi and Yamanaka, 2006; Tsuji et al., 2010). Compared to iPSCs from human and mice, a relatively small number of nonhuman primate iPSC lines exist (Wu et al., 2012). However, large animals will be dominant in ironing out clinical safety issues from reprogramming technology. Viral reprogramming facilitates the threat of insertional mutagenesis (Amabile and Meissner, 2009; Takahashi et al., 2007), and the transcription factors used for reprogramming appear to have an oncogenic character and are suspected to cause cancer if not fully silenced (Takahashi et al., 2007; Takahashi and Yamanaka, 2006). Thus, advanced reprogramming technology, common quality criteria of iPSCs, and preclinical studies in nonhuman primate models are in our opinion of foremost interest.
In recent studies, only cells from fetal skin and liver have been used in marmoset iPSC generation. Furthermore, a mixture of four retroviruses was transduced in fetal skin fibroblasts for the generation of iPSCs, and retrovirus-mediated transduction of six human pluripotency markers was performed in fetal liver cells (Tomioka et al., 2010; Wu et al., 2010). Attempts to reprogram with cells derived from the bone marrow were suspected not to be true iPSCs (Tomioka et al., 2010). Until now, fully reprogrammed iPSCs exist from fetal skin fibroblasts and fetal hepatocytes, facilitating the expression of endogenous pluripotency markers and the complete silencing of the introduced transgenes (Tomioka et al., 2010; Wu et al., 2010).
In this study, we used adult bone marrow–derived somatic cells of our nonhuman primate model for the generation of iPSCs. The cells were transduced with a novel, advanced, and excisable quad-cistronic lentiviral vector system (Voelkel et al., 2010; Warlich et al., 2011). With respect to later use in transplantation, derivation of differentiated cell types and plasticity were tested thoroughly.
Material and Methods
Animal rights declaration
All animal work was performed strictly according to institutional guidelines and national regulations and was approved by the state office for protection of nature, environment, and consumers (LANUV) of North-Rhine Westphalia, Germany. Retrieval of MSCs from the bone marrow was performed only in the context of other veterinary measures and was approved by the Institutional Animal Care. The teratoma assays in immunodeficient NOD.Cg-PRKDCscidIL2rgpm1Wjl/PRKDC mice were performed at the Institute for Laboratory Animal Science and Central Animal Facility, Hannover Medical School, Hannover, Germany (animal license 10/0209).
Generation of marmoset iPSCs via a lentiviral vector system
For the generation of iPSC lines, bone marrow–derived mesenchymal stem cells (MSCs) were obtained from 3 different animals (age 3–5 years). The cells were isolated as described before (Bernemann et al., 2011), seeded in 10-cm petri dishes (Greiner Bio-One GmbH, Frickenhausen, Germany) in MSC medium [Dulbecco's modified Eagle medium (DMEM; Lonza)+15% fetal calf serum (FCS; Lonza)+1% sodium pyruvate, c-c-pro+1% penicillin/streptomycin, c-c-pro+0.1% ascorbic acid] and cultivated until confluence.
For reprogramming, 3.5×105 cells were seeded on a six-well plate coated with Matrigel (BD-Bioscience, Heidelberg, Germany) 1 day before transduction and cultivated at 37°C and 5% CO2. The medium was replaced by fresh MSC medium supplemented with protamine sulfate (8 μg/mL), and the cells were incubated for 30 min at 37°C and 5% CO2 prior to transduction. Afterward, lentivirus-containing supernatant with a multiplicity of infection (moi) of 0.5 was added to the medium. As described before, the lentivirus contained a spleen focus-forming virus (SFFV)-driven quad-cistronic vector with a dTomatoRed reporter gene including human pluripotency markers OCT3/4, KLF4, SOX2, and C-MYC and flanked by FLP sites to ensure the later removal of the transgenes (Voelkel et al., 2010; Warlich et al., 2011). The cells were cultivated in MSC medium supplemented with valproic acid (VPA, 2 mM). After the detection of a fluorescent signal on days 5–10, all dTomatoRed-positive cells were sorted and seeded on a Matrigel-coated plate. To adapt the cells to new medium, the day after sorting the cells were cultivated in MSC medium mixed 1:1 with mTeSR (StemCell Technologies SARL, Grenoble, France). At 24 h later, only mTeSR with the small molecules PD0325901 (1 μM), SB431542 (2 μM), ascorbic acid (20 μg/mL), and Thiazovivin (TAV) (10 μM) (Selleck Chemicals Co. Limited, Munich, Germany) was used until colony formation on days 7–12. The colonies were transferred to 30 g γ-irradiated CD1 mouse embryonic fibroblast (MEF) feeder layer and cultivated in human embryonic stem cell medium [hESM; Knockout DMEM/F12 (Gibco)+20% Knockout Serum Replacement (KSR; Gibco)+1% minimum essential medium (MEM) (nonessential amino acids, NEAA; Gibco)+1% penicillin/streptomycin, c-c-pro+0.5%
Reverse transcriptase PCR
Total RNA was extracted from iPSCs or teratomas using RNeasy Mini Kit (Qiagen®, Hilden, Germany) following the manufacturer's protocol. Contaminant DNA was removed by DNase treatment using the RNase-free DNase Set (Qiagen®, Hilden, Germany). In general, 1 μg of RNA was used for reverse transcription with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). By adding Oligo(dT) primers (TIB Molbiol, Berlin, Germany), only mRNA was transcribed. For the analysis of the pluripotency markers, a PCR reaction of 30 μL per sample was set up as follows: 24 μL dH2O, 3 μ 1×PCR buffer (NEB, Frankfurt, Germany), 0.5 Units of Taq Polymerase (NEB, Frankfurt, Germany), 100 mM dNTPs (Fermentas, St. Leon-Rot, Germany), 20 pmol/μL of each primer, and 1 μg of cDNA. Cycling conditions contained a precycling step at 95°C for 3 min followed by 35 cycles of denaturation at 95°C for 45 sec, annealing at 58°C for 45 sec, and extension at 72°C for 90 sec, with a final extension step at 72°C for 10 min. Sequencing of cDNA fragments of the expected size was performed in house using a BigDye™ Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems v1.1) according to the manufacturer's instructions (Sanger) in 96-well PCR-plates (Kisker, Steinfurt, Germany) in a C1000 Thermal Cycler (BioRad, München, Germany). A summary of oligonucleotide sequences, fragment sizes and PCR conditions is enlisted in Table 1.
Asterisks indicate sequence was anticipated from human homolog.
IgG, immunoglobulin G; IgM, immunoglobulin M.
Alkaline phosphatase staining
For alkaline phosphatase (AP) staining, the AP staining kit (Dako EnVision™ G2 System/AP, Rabbit/Mouse (Permanent Red), K5355) was used according to the manufacturer's instructions. The iPSCs were grown on glass slides, and after 4 days washed with phosphate-buffered saline (PBS) and incubated with substrate working solution of Permanent Red Chromogen for 5 min at 37°C. The slides were then rinsed with deionized water and further analyzed under a Keyence Biozero microscope (Keyence Germany GmbH, Neu-Isenburg, Germany).
Immunofluorescence staining
For detection of proteins in nonhuman primate cell cultures and tissues, human directed antibodies are mostly specific. In the case of biologically artificial iPSCs and embryonic stem cells (ESCs), proteins might be produced randomly, and it is difficult to judge specificity by histology in this context. Therefore, in our group, a large tissue and organ bank of more than 20 animals and 36 tissues exists to test specificity and to provide negative control samples of all antibodies used in immunohistochemistry and immunofluorescence in this study.
For the examination of pluripotency markers, the polyclonal iPSC line was expanded in a 12-well plate (Greiner Bio-One GmbH, Frickenhausen, Germany) on MEFs, fixed with 4% paraformaldehyde (PFA), and kept in PBS until use. Before incubation with the primary antibody, unspecific binding was minimized by treatment with bovine serum albumin (BSA). The cells were washed twice with PBS. The primary antibodies used in the study are listed with dilutions and companies, respectively, in Table 2. In general, antibodies were diluted in PBS+0.1% Triton X-100 and kept on cells overnight at 4°C. After incubation with the primary antibody, the cells were washed twice with PBS. The corresponding secondary antibody linked to Alexa dye A488 or A568 was diluted in PBS+0.1% Triton X-100 in an appropriate dilution, and it was incubated on the cells at room temperature for 60 min. Afterward, the cells were washed with PBS again, and the nuclei were stained with Hoechst (1:10,000) for 5 min at room temperature. The cells were analyzed using the fluorescence microscope Olympus CKIX71 (Olympus, Hamburg, Germany).
Embryoid body formation
For embryoid body (EB) formation, undifferentiated iPSCs were treated with collagenase IV (1 mg/mL, 4 min at 37°C) to detach the colonies from MEFs. Colonies were centrifuged at 200×g for 3 min in a tabletop centrifuge, and the pellet was resuspended softly (DMEM, 10% FBS, 1% sodium pyruvate, and 1% antibiotic/antimycotic) but not dissociated by force and cultured on 0.5% gelatin-coated (Sigma Aldrich, Steinheim, Germany) plates (Greiner Bio-One GmbH, Frickenhausen, Germany) for 20 days.
Teratoma assay
To study the differentiation capability of the iPSCs obtained, a cell suspension of 1.5×106 cells was injected into the kidney capsule of immunodeficient NOD.Cg-PRKDCscidIL2rgpm1Wjl/PRKDC mice. The tumors were removed from the mice 6–8 weeks after injection and were fixed in 4% PFA for histology. Parts of the tumors were frozen in liquid nitrogen, and RNA extraction was performed as described above.
Karyotyping
Karyotype analyses were performed of the obtained iPSC line from bone marrow–derived cells from the common marmoset. Metaphases were prepared according to standard procedures. Fluorescence R-banding using chromomycin A3 and Methyl Green was performed as described in detail earlier (Schlegelberger et al., 1999). Karyotypes were analyzed using the marmoset chromosome map described previously (Sherlock et al., 1996).
Differentiation into ectodermal cell type
The differentiation of iPSC clusters into neural progenitor cells was induced by DMEM-F12 (PAA, Coelbe, Germany)/Neurobasal Medium (Invitrogen, Darmstadt, Germany) (50:50) supplemented with 1:100 N2 supplement (Invitrogen, Darmstadt, Germany), 1:50 B27 supplement (Invitrogen, Darmstadt, Germany), 5 ng/mL epidermal growth factor (EGF; PeproTech, Hamburg, Germany), 5 ng/mL fibroblast growth factor-2 (FGF-2; PeproTech, Hamburg, Germany) with 1% penicillin/streptomycin/glutamine on polyornithine-laminin–coated dishes. After 3–5 days, the medium was replaced by neurobasal medium supplemented with 1:100 N2 supplement and 1:50 B27 supplement with 1% penicillin/streptomycin/glutamine. The medium was changed every 2–3 days. After 2 weeks, cells and remaining colonies were gently dissociated using 0.05% trypsin in PBS and plated on Matrigel-coated (hESC-qualified matrix, BD Biosciences, Heidelberg, Germany) glass coverslips for staining. Matrigel was diluted in neurobasal medium, and coating was performed over night at room temeprature. To verify neural differentiation, immunofluorescence staining with antibodies against the neural markers SOX1, PAX6, and Tubb3 was conducted (Table 2).
Differentiation into definitive endoderm
The endodermal differentiation protocol was performed as published before (D'Amour et al., 2005) with minor modifications. iPSC colonies were removed from feeder cells via collagenase treatment and passaged into 12-well dishes (Greiner Bio-One GmbH, Frickenhausen, Germany) at a density of 3×105 cells/well coated with Matrigel (BD-Bioscience, Heidelberg, Germany) and cultivated for 6–7 days prior to differentiation with mTeSR (StemCell Technologies SARL, Grenoble, France) in the presence of 30 ng/mL of bFGF (PeproTech, Hamburg, Germany). Medium for directed differentiation contained RPMI, 1% GlutaMAX, and 1% antibiotic–antimycotic. On day 1 of differentiation, 100 ng/mL Activin A (PeproTech, Hamburg, Germany) and 25 ng/mL Wnt3a (PeproTech, Hamburg, Germany) were added to the medium. On the second day, medium was changed to 100 ng/mL Activin A, 25 ng/mL Wnt3a, and 0.2 % FCS (Lonza, Basel, Switzerland). From day 3 until day 6, the medium contained 100 ng/mL Activin A and 0.2% FCS.
Differentiation into mesenchymal cell types
The differentiation protocols for adipocytes, osteocytes, and chondrocytes of the marmoset were described before (Bernemann et al., 2011). Briefly, the chondrogenic differentiation of iPSCs was induced by using chondrogenic differentiation medium (0.1 μM dexamethasone, 1 mM sodium pyruvate, 0.17 mM
Adipogenic differentiation was induced by the addition of 1 M dexamethasone, 0.2 mM indomethacin, 0.5 mM IBMX, and 10 μg/mL insulin in DMEM with 10% FBS for 3 days. The medium was replaced by DMEM, 10% FBS, and insulin (20 g/mL) for 2 days. The cultivation procedure was repeated four times before cells were stained with Oil Red O dye to detect the generation of fat vacuoles.
For osteogenic differentiation, the cultivation medium DMEM+15% FBS was supplemented with 0.1 M dexamethasone, 10 mM β-glycerophosphate, 0.05 mM
Differentiation into megakaryocytes was performed in 12-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany) with 3×105 cells in the presence of 2 mL of IMDM (Lonza, Basel, Switzerland), 5% FCS, 100 ng/mL Flt3-L (PeproTech, Hamburg, Germany), 100 ng/mL interleukin-3 (IL-3; PeproTech, Hamburg, Germany), and 50 ng/mL thrombopoietin (TPO; PeproTech, Hamburg, Germany) for 2 days. On day 3, culture medium was changed to 2 mL of Iscove's modified Dulbecco medium (IMDM), 5% FCS, 100 ng/mL Flt3-L, and 100 ng/mL TPO until day 5. In the next 10 days, attached cells displayed the typical megakaryocytic morphology and expressed diverse typical marker genes in RT-PCR.
Results
Generation of marmoset iPSCs from bone marrow–derived MSCs
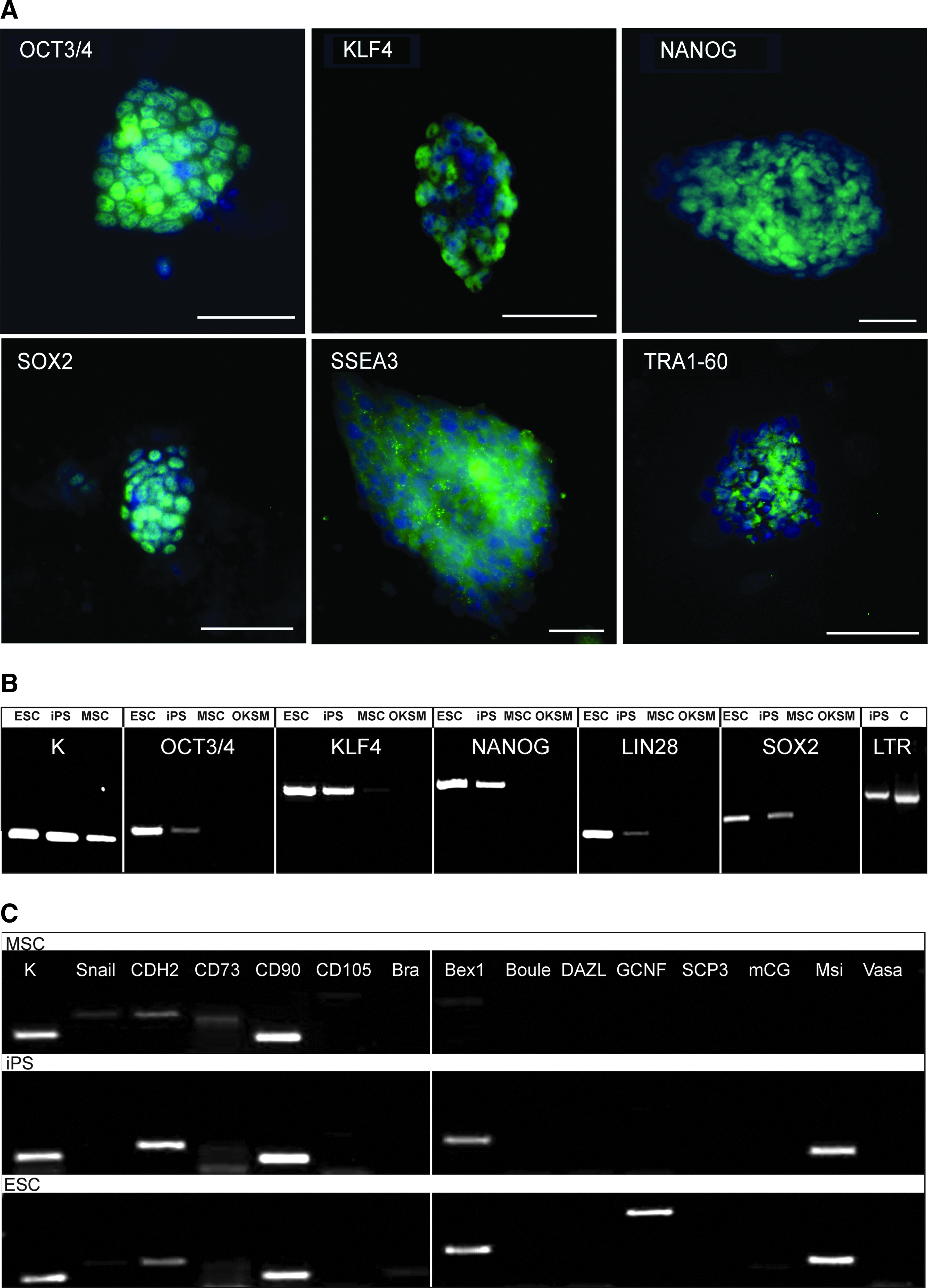
Bone marrow–derived cells from the common marmoset were transduced with a lentiviral construct containing the human pluripotency markers OCT3/4, KLF4, SOX2, and C-MYC (OKSM). A moi of 0.5 was used for the transduction. All dTomatoRed-positive cells were sorted after a fluorescent signal could be detected. Colony formation could be observed about 1 week after sorting with typical ESC-like morphology. Of 7×105 dTomatoRed-positive cells, we obtained 278 colonies (reprogramming rate >0.04%). The colonies were picked and cultivated on Matrigel for few passages before they were transferred onto irradiated MEFs. After passage 20, the cells developed a flat and more mouse ESC-like morphology (Fig. 1A) and strong AP signal (Fig. 1B) that could be confirmed by RT-PCR and AP staining. ESCs showed weaker expression of AP, and MSCs showed none (Fig. 1B). The iPSCs obtained showed a doubling rate of 24 h (data not shown). All characterization and differentiation experiments were performed with a polyclonal iPSC line generated with selected colonies from the original culture by morphology, chromosomal integrity (46, XX karyotype) (Fig. 1C), and strong NANOG signal (confirmed by RT-PCR).

Generation of iPSCs. Bone marrow–derived mesenchymal cells were used for reprogramming. (
Detection of endogenous pluripotency markers by RT-PCR
For the confirmation of the reprogramming status of the obtained iPSCs, the gene expression of the pluripotency markers was analyzed by immunofluorescence (OCT3/4, KLF4, SOX2, NANOG, SSEA-3, TRA-1-60; Fig. 2A) and RT-PCR (OCT3/4, KLF4, SOX2, NANOG, and LIN28) (Fig. 2B, composite display) with marmoset-specific primers (Table 1). The analysis showed gene expression of endogenous OCT3/4, KLF4, and SOX2; the transgenes could not be detected in the iPSCs. Additionally, endogenous NANOG and LIN28 expression could be shown in the iPSCs. All genes could be easily distinguished from marmoset sequences due to codon optimization of the human transgenes; however, the only transgene with unchanged sequence and 100% similarity with the marmoset was C-MYC, so it was not included in Fig 2. These results show that the introduced pluripotency markers were silenced in the obtained iPSCs, whereas endogenous expression was activated. Furthermore, RT-PCR of 14 selected genes (Fig. 2C, composite display) displayed the presence of the epithelial–mesenchymal transition-associated marker CDH2, mesenchymal stem cell marker CD90, the presence of early embryonic gene Bex1 in iPSCs and ESCs, but the absence of germ cell nuclear factor (GCNF) in MSCs and iPSCs (K=housekeeping gene, OKSM=vector).
(
Undirected differentiation: Embryoid body and teratoma formation
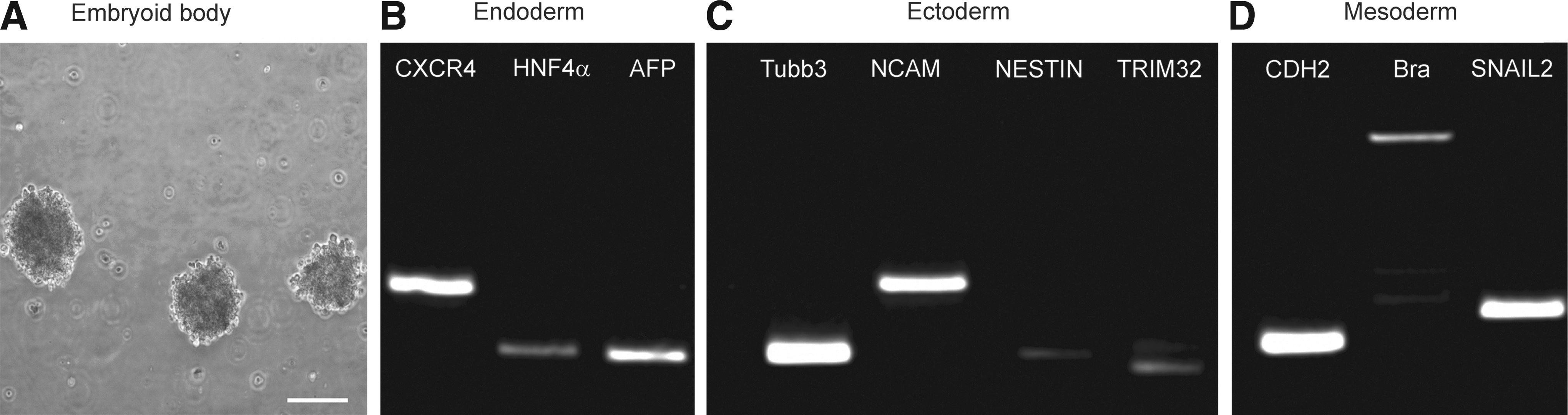
To examine the spontaneous differentiation of the generated iPSCs, an EB culture was prepared. The iPSCs were kept in suspension culture in gelatin-coated petri dishes for 20 days (Fig. 3A). To analyze the gene expression in the formed EBs, RT-PCR was performed via marmoset-specific primers (Table 1). We could show that the generated iPSCs have the potential to differentiate into all three germ layers. For endodermal cells, CXCR4, HNF4α, and AFP were chosen as markers (Fig. 3B); ectodermal development could be shown by strong Tubb3 and NCAM expression and weaker NESTIN and TRIM32 signals (Fig. 3C). Mesodermal development displayed CDH2, brachyury, and SNAIL2 gene expression (Fig. 3D).
(
Teratoma formation
To study the capability of the marmoset iPSCs in more detail, 1.5×106 iPSCs were injected into kidney capsules of immunodeficient NOD.Cg-PRKDCscidIL2rgpm1Wjl/PRKDC mice. Three weeks after injection, tumors were palpable in 4 of 5 mice. Six weeks after injection, the tumors were removed. Gross morphology showed solid tumor masses with frequent fluid-filled cavities of varying size and grafting into the renal tissue of the host mice. Markers of ectodermal, mesodermal, and endodermal tissues were analyzed by immunohistochemistry staining, by histology and via RT-PCR with typical marker genes (Fig. 4). In immunohistochemistry staining, a specific FOXA2 signal for endodermal, brachyury for mesodermal, and Tubb3 for ectodermal development could be detected (Fig. 4A–C). In Hematoxylin & Eosin staining, mesodermal tissue was demonstrated by formation of cartilage (Fig. 4D). Ectodermal tissue represented by capillaries and neuronal cells showing formation of immature rosettes could also be found (Fig. 4E). Endodermal structures were represented by gland-like structures lined by cuboidal epithelium with cilia (Fig. 4F). Furthermore, RT-PCR reaction of teratoma tissue (Fig. 4G, composite figure) displayed typical genes (Table 1) for endodermal (GATA4+, CXCR4+, HNFa−, AFP+), ectodermal (Tubb3+, NCAM+, TH+, TRIM32+, NURR+, NESTIN+), and mesodermal cell types (CDH2+, Bra+, SNAIL2+).

Teratoma formation in kidney capsule of immunodeficient NOD.Cg-PRKDCscidIL2rgpm1Wjl/PRKDC mice. (
Directed differentiation of iPSCs
To evaluate the potential of the marmoset iPSCs obtained, these iPSCs were differentiated into mesodermal cells like adipocytes, chondrocytes, and osteocytes. Differentiation followed established protocols (Bernemann et al., 2011) and showed clear evidence of adipocyte formation in which the lipid granules could be visualized via Oil Red O staining (Fig. 5A). Calcification in early osteogenic differentiation was observed by von Kossa staining (Fig. 5B) and chondrocytes by Alcian Blue staining (data not shown). Upon differentiation in the hematopoietic direction, morphological characteristics of megakaryocyte formation could be seen (Fig. 5C–F). Following protocols for differentiation into the neural lineage, typical markers like SOX1 (Fig. 5G), PAX6 (Fig. 5H), and Tubb3 (Fig. 5I) were detectable via immunofluorescence staining. Early definitive endoderm could be seen using the protocols of D'Amour et al. (2005). For the characterization of endodermal cell types, the liver-specific marker SOX17 (Fig. 5J) as well as FOXA2 (Fig. 5K), which is expressed in most of the organs after 8 days of Activin A stimulation (Fig. 5L, merged picture), were used.

Directed differentiation into adipocytes (Oil Red O staining) (
Furthermore, typical genes related to the cell types generated could be found by RT-PCR reaction (Fig. 5M, composite figure). Successful mesodermal differentiation could be demonstrated via high expression of the chondrogenic marker CYR61 and the two osteogenic markers RUNX2 and MMP2, as well as the hematopoietic markers CD37 and ITGB3b. After neural induction, the neural markers NURR, NESTIN, PAX6, SOX1, Musashi, and TH could be detected via RT-PCR (Englund et al., 2005; Walcott and Provis, 2003; Wang et al., 2011). Last, but not least, endodermal cells showed high expression of CXCR4, SOX17, and FOXA2.
Discussion
The marmoset is an important nonhuman primate model in regenerative medicine because it is the only primate model with the triptych of transgenic germ-line transmission and established ESCs and iPSC as experimental tools. Currently, there is much debate about retrieval of somatic cells and quality of iPSCs with respect to final application in human therapy.
In this study, we presented an advanced system for the production of nonhuman primate iPSCs. The quad-cistronic and extractable lentiviral vector system was transduced with a very low moi of 0.5 to avoid insertional mutagenesis. The sequences of the key reprogramming genes (OCT3/4, KLF4, SOX2, and C-MYC) were human-based, codon-optimized, and used in the presence of a cocktail of small molecules during iPSC generation to enhance reprogramming efficiency. Furthermore, in comparison to published data, we used an adult somatic cell type for reprogramming.
The iPSC colonies sustained an undifferentiated state after 64 passages expressing the endogenous pluripotency markers OCT3/4, KLF4, SOX2, C-MYC, NANOG, and LIN28, whereas the introduced transgene cassette was silenced and, in addition, excised to avoid unintentional reactivation. The potential of the polyclonal cell line to differentiate into all three germ layers was demonstrated by EB and teratoma formation and directed differentiation in various cell types.
Interestingly, some of the MSC-derived iPSC colonies displayed a more mouse ESC-like morphology, similar to reports of Tomioka et al. (2010), and different from other marmoset ESC lines (Fleischmann et al., 2009; Muller et al., 2009; Sasaki et al., 2005). This was observed independently from silencing and extraction of the transgenes. During the long period of cultivation, the obtained iPSCs also showed a human ESC-like shape. Similar phenomena could be observed in different culture methods of human iPSCs by others (Hanna et al., 2010; Tesar et al., 2007), but in our case the addition of bFGF in human or standard mouse ESC culture medium did not stabilize the colonies' phenotype to the one or the other side.
Furthermore, from our observation in marmoset iPSC production, small molecules seem to have a great positive influence, as reported by others (Choi and Nam, 2012; Laping et al., 2002). However, 2i and 3i inhibitor treatment (Li et al., 2009) showed no improvement in early iPSC colony formation (data not shown).
Until now, there has been no generally accepted molecular marker to indicate the fully reprogrammed state of iPSCs. Even the presence of ALP, NANOG, and LIN28 is debated to date (Hanna et al., 2010; Wang et al., 2010). However, in our study we could show that the obtained polyclonal iPScline was strongly AP positive and LIN28 and NANOG expression was reactivated, corroborating true reprogramming (Wang et al., 2012).
As an additional contribution to our characterization and differentiation experiments, we evaluated whether the obtained iPSCs maintained closer genetic characteristics to MSCs or to ESCs. In a comparative RT-PCR with 14 selected genes, ESCs express some MSCs and epithelial–mesenchymal transition-related markers like CDH2 and CD90 (Nakajima et al., 2004; Ock et al., 2010), whereas undifferentiated ESCs and the obtained iPSC line are deficient of brachyury, Boule, DAZL, VASA, mCG, and SCP3 expression (Muller et al., 2009). In addition, we detected expression of the early embryonic gene Bex1 (Ruau et al., 2008; Tomioka et al., 2010), but, as expected, the late GCNF was just present in ESCs. GCNF is involved in terminal differentiation in spermatogenesis and is therefore the only difference in our gene selection between ESCs and iPSCs. In undirected differentiation and teratoma formation, all three germ layers could be generated and confirmed by RT-PCR and immunohistochemistry, which demonstrates a high quality of the marmoset iPSC line.
Furthermore, the cellular iPSC plasticity in vitro was excellent: Adipocytes, chondrocytes, osteogenic cells, megakaryocytes, neural progenitors, and definitive endoderm were generated with standard protocols developed for human ESCs.
In the original unselected iPSC culture, karyotyping revealed a trisomy most likely of chromosome 6. Until now, the meaning of a trisomy 6 in the marmoset is unclear. However, in humans, trisomy 6 is a recurrent aberration of neoplasia, e.g., acute lymphoblastic leukemia. Aneuploidies are well known to occur in murine and human iPSC lines, and possible causes could be the use of reprogramming factors (Baum et al., 2011) or the histone deacetylase (HDAC) inhibitor VPA (Sakurada, 2010). Chromosomal aberrations indicate once again the necessity to use a low virus titer for transduction, excisable constructs, and lines carefully monitored for chromosomal abnormalities prior to therapeutic usage.
Future projects comprise cardiomyocyte and granulocyte production, autologous transplantation, and the reduction of allogenicity by repression of the major histocompatibility complex (MHC) class 1 gene for in vivo cell replacement therapy. Further problems to be taken care of are avoidance of integration mutagenesis by episomal or RNA-facilitated episomal reprogramming and abrogation of teratoma formation. In terms of iPSC generation, this small New World primate proves once again to be a valuable link between rodents and humans and a reasonable and cost-efficient preclinical model for regenerative medicine.
Footnotes
Acknowledgments
We thank Kirsten Elger and Michael Engelmann for excellent technical support. Our gratitude also includes Prof. Stefan Schlatt from the Centre for Reproductive Biology and Andrology (CeRA) for providing us with material of the common marmoset and Dr. S. Fuchs from the Institute for Human Genetics, University Medical Centre Hamburg-Eppendorf. This work is supported by funding from the German Research Foundation (DFG) for the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy) and from the Institute for Transfusion Medicine of the Hannover Medical School.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.