
Abstract
Abstract
Pluripotent stem cells (PSCs) have the potential to differentiate into many cell types and therefore can be a valuable source for cell therapy. Embryonic stem cells (ESCs), which are derived from the inner cell mass (ICM) of the blastocyst, are representative of PSCs. However, use of these cells has some limitations, especially ethical restrictions and immune response. As a result, researchers have been looking for other cell sources or strategies to overcome these limitations. One kind of cellular reprogramming is the process of guiding mature cells into a state of gene expression similar to PSCs. It has been demonstrated that somatic cells can be reprogrammed by various methods, including somatic cell nuclear transfer (SCNT) and cell fusion with ESCs or treatment with their extracts. This implies that some factors in oocytes and ESCs are able to initiate the reprogramming process. Accordingly, induced pluripotent stem cells (iPSCs) have been derived from somatic cells by ectopic expression of some transcription factors. This discovery has resulted in raising several important questions about the mechanisms by which these factors influence the reprogramming and epigenetic status of the cells. iPSCs hold great promise for regenerative medicine, developmental biology, and drug discovery because they circumvent problems associated with both ethical issues and immunological rejection. Here we review the experiments involved in the discovery of iPSCs, important factors in their reprogramming, and their future perspectives in cell therapy.
Introduction
Nuclear Reprogramming
Because mature cells are genetically the same as early embryonic cells, and the epigenetic changes are reversible (Briggs and King, 1952; Hochedlinger and Jaenisch, 2002; Wilmut et al., 1997), it should be possible to revert somatic cells to an embryonic state, so called nuclear reprogramming (Briggs and King, 1952). The first demonstration of nuclear reprogramming by transfer of a somatic cell nucleus into an enucleated oocyte, somatic cell nuclear transfer (SCNT), occurred in the 1950s. This involved transferring the nuclei of Xenopus blastula cells into oocytes (Briggs and King, 1952). This finding and subsequent experiments by Gurdon and colleagues indicated that cell specialization does not involve any loss, irreversible inactivation, or permanent change in genes required for development (Gurdon, 1962; Gurdon et al., 1975).
In the intervening years, cloned animals and ESC lines from cloned embryos have been established in many species (Hochedlinger and Jaenisch, 2002; Inoue et al., 2005; Jaenisch and Young, 2008; Wilmut et al., 1997). SCNT experiments and subsequent findings clearly demonstrated that epigenetic changes during cellular differentiation are reversible and somatic cells could be reprogrammed to a pluripotent state by defined factors (Muller et al., 2009). In 2002, the ESCs derived from cloned embryos were differentiated to hematopoietic cells, producing the hope of creating patient-specific cells for therapeutic purposes (Rideout et al., 2002). However, besides being technically challenging, SCNT has some limitations, including extremely low efficiency, severe developmental abnormalities in cloned animals, and ethical problems concerning the use of human oocytes (Gurdon et al., 2003; Wakayama and Yanagimachi, 1999).
An alternative approach to reprogramming that was adopted in 1970s was the use of cell fusion (Foshay et al., 2012; Miller and Ruddle, 1976, 1977; Taranger et al., 2005). These approaches also manifest some deficiencies; for example, the fusion rate is low and the hybrid cells after fusion have two sets of chromosomes, which limits the use of these cells for clinical applications (Brinster, 1974; Stewart and Mintz, 1982; Tada et al., 2001). Another related approach was the use of extracts from pluripotent cells to reprogram somatic nuclei. This strategy seems to have resulted in some success in the reversion of several aspects of cell differentiation (Shuang and EnKui, 2008).
Both fusion with stem cells and treatment with their extracts are dependent on the presence of pluripotent cells that provide the microenvironment needed for reprogramming. The reprogramming efficiencies of these methods are poor, and their applications are limited by various elements (Patel and Yang, 2010; Shuang and EnKui, 2008). In contrast, the new approach of reprogramming by exogenous expression of pluripotency-related genes has overcome many problems associated with former methods (Shuang and EnKui, 2008).
One of the most important findings that helped researchers to create induced pluripotent stem cells (iPSCs) was the fact that ectopic expression of lineage-associated transcription factors can result to change cell fate. For example, it was shown that the transcription factor MyoD can induce muscle-specific properties in fibroblasts (Davis et al., 1987). Graf showed that upon overexpression of the myeloid transcription factor C/EBPα in primary B and T cells, they could be converted into functional macrophage-like cells (Xie et al., 2004), and ectopic expression of interleukin-2 (IL-2) and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors could lead to myeloid conversion in myeloid progenitor cells (Kondo et al., 2000).
Induced Pluripotency
On the basis of previous studies, it was known that pluripotent stem cells (PSCs) have the potential to reprogram somatic cells. Therefore, researchers speculated that these cells may contain some trans-acting factors for induction of reprogramming (Cui et al., 2009). Given this possibility, Takahashi and Yamanaka examined 24 candidate pluripotency-related factors to investigate their ability to induce pluripotency in differentiated somatic cells. The 24 candidates included transcription factors essential for pluripotency in ESCs and early embryos, such as: Oct4, Sox2, and Nanog; products of (proto)oncogenes important for ESC proliferation, such as Tcl1, Stat3 and c-Myc; and some less well-known factors specifically expressed in ESCs, such as ECAT1, Esg1, Fbx15, and Klf4 (Takahashi and Yamanaka, 2006).
Transfection of all 24 candidate genes resulted in Fbx15-positive colonies, in stark contrast to single-gene transfections, which yielded no Fbx15-positive colonies. Systematic reduction in the number of factors used in the transfections led to a 10-factor combination with improved Fbx15-positive colony formation over the 24-factor transfections. Four factors—Oct4, Sox2, c-Myc, and Klf4—were identified in the 10-factor combination, whose removal significantly compromised colony formation (Takahashi and Yamanaka, 2006).
The cells derived were not exactly identical to mouse ESCs. For instance, these reprogrammed cells expressed markers of pluripotency such as stage-specific embryonic antigen 1 (SSEA-1) and Nanog and they generated teratomas, but they expressed lower levels of several key pluripotency genes compared with ESCs, had incomplete promoter demethylation of stem cell regulators such as Oct4, failed to generate postnatal chimeras when injected into preimplantation mouse embryos, failed to contribute to normal development, and therefore failed the most important test for pluripotency (Takahashi and Yamanaka, 2006). So, while these iPSCs seemed to be only partially reprogrammed, they were very close phenotypically to conventional ESCs. Several laboratories independently were able to reproduce and improve upon these findings. These laboratories used selection markers based on pluripotency factors like Nanog or Oct4 expression and made great improvements in the quality of generated iPSCs. Stable cell lines were derived that morphologically and functionally were more closely resembling ESCs in transcription, imprinting, and chromatin-modification profiles (Maherali et al., 2007; Meissner et al., 2007; Okita et al., 2007). These three groups generated chimeras from iPSCs and demonstrated their germ-line contribution. In 2007, another group reported the derivation of iPSCs from somatic cells based on morphological criteria instead of any reporter selection, although this method needs careful characterization of the resultant cell lines (Qin et al., 2007). This approach is most powerful when combined with, for example, doxycycline-inducible vectors, because cells that have entered a self-sustaining pluripotent state can be easily selected by removal of doxycycline (Stadtfeld et al., 2008a).
Yamanaka's group reported that human iPSCs were obtained from human skin cells, using similar morphological selection strategies. They reprogrammed adult human dermal fibroblasts with the same four factors used to reprogram mouse fibroblasts (Takahashi et al., 2007). An obvious caveat to the eventual use of iPSCs in a clinical setting is the presence of c-Myc in the Yamanaka reprogramming cocktail. Omission of c-Myc from the Yamanaka reprogramming cocktail reduced the efficiency of iPSC generation (Nakagawa et al., 2008).
A second group used a different combination of four crucial factors—OCT4, SOX2, NANOG, and LIN28—to reprogram human somatic cells (Yu et al., 2007). The work described in Yu et al. (2007) was based on gene expression profiles of somatic cell hybrids obtained from the fusion of human ESCs to myeloid precursors. The reprogrammed myeloid cells were enriched for genes involved in pluripotency and this eventually led to the identification of OCT4, SOX2 (also used by Yamanaka), NANOG, and LIN28 as critical reprogramming factors (Yu et al., 2007).
Recently, it was shown that iPSCs are even capable of generating mice upon injection into tetraploid blastocysts, suggesting that at least some iPSC clones have a developmental potency equivalent to ESCs (Stadtfeld et al., 2010).
iPSCs have been derived from different species, including rat (Li et al., 2009), mouse (Aoi et al., 2008; Carey et al., 2009; Okita et al., 2008; Takahashi and Yamanaka, 2006), human (Aoki et al., 2010; Takahashi et al., 2007; Yu et al., 2007), rhesus monkey (Liu et al., 2008), marmoset (Wu et al., 2010), sheep (Li et al., 2011; Liu et al., 2012), pig (Esteban et al., 2009; Montserrat et al., 2012), horse (Nagy et al., 2011), goat (Ren et al., 2011), bovine (Han et al., 2011), and different somatic cells such as skin fibroblast cells (Takahashi et al., 2007; Takahashi and Yamanaka, 2006), neural cells (Eminli et al., 2008), stomach and liver cells (Aoi et al., 2008), keratinocytes (Maherali et al., 2008), mouse adipose tissue-derived and neural stem cells (Tat et al., 2010), melanocytes (Utikal et al., 2009), neural stem cells (Kim et al., 2009b), amniotic fluid cells (Galende et al., 2010), cord blood cells (Giorgetti et al., 2009), and pancreatic β-cells (Stadtfeld et al., 2008a), demonstrating that basic characteristics of pluripotency and transcriptional network remain conserved during evolution and development.
Different Factor Delivery Systems
The very low reprogramming efficiency (typically less than 0.1%) and the unsuitability of integrating viral vectors as a delivery system for cells destined for clinical use has led to a large effort to improve efficiency and use more clinically compatible delivery systems. Various approaches have been used to transfer reprogramming factors into somatic cells, with different efficacy and quality of resultant iPSCs. The gene delivery approaches can be divided into the categories of: (1) integrating vectors, (2) nonintegrating vectors, (3) integrating vectors, which can be removed after integration into the genome, and (4) nonnucleic acid–based approaches. Different methods used for producing iPSCs are summarized in Table 1.
Generation of iPSCs with Integrating Vectors
The original iPSC generation study used retroviral vectors that constitutively express the reprogramming factors (Takahashi and Yamanaka, 2006). The constitutive nature of the promoters used in these vectors means that obtaining iPSCs that no longer express the transgenes relies on the reprogrammed cells silencing the introduced vectors. Alternative lentiviral vectors are more efficient than retroviral vectors in infecting different somatic cell types and can express polycistronic cassettes encoding all required genes, therefore increasing the reprogramming efficiency (Carey et al., 2009). Both retrovrial and lentiviral vectors, however, are incompletely silenced. Lentiviral vectors were less efficiently silenced than retroviral vectors and thus may inhibit differentiation (Brambrink et al., 2008). The use of inducible lentiviral vectors, whose expression can be controlled by, for example, doxycycline, eliminated the negative effects of unnecessary expression of the transgenes and therefore resulted in producing completely reprogrammed cells (Hamilton et al., 2009). In 2011, it was shown that expression of only the miR302/367 cluster by lentiviral vectors, without the use of exogenous transcription factors, rapidly and efficiently reprogrammed mouse and human fibroblast cells to an iPSC state. This microRNA (miRNA)-based reprogramming approach was twice as efficient compared to standard transcription factor delivery methods using Oct4, Sox2, Klf4, and c-Myc (Anokye-Danso et al., 2011).
Generation of iPSCs without Integration of Transgenes
Generating the iPSCs using retroviral and lentiviral vectors causes a random integration of the desired genes into the target genome, which can result in unexpected silencing of necessary genes or undesired activation of loci. Therefore, other methods of delivery that are safer and nonintegrating have been investigated.
Adenoviruses have been successfully used to generate iPSCs with DNA demethylation patterns characteristic of reprogrammed cells, expression of endogenous pluripotency factors, teratoma-forming ability, and contribution to multiple tissues, including the germ line in chimeric mice (Stadtfeld et al., 2008b). Adenoviral vectors have also been used for reprogramming human fibroblasts into iPSCs (Zhou and Freed, 2009). iPSC generation using Sendai virus, a nonintegrating RNA virus, without risk of altering the host genome, is an efficient strategy for generating safe iPSCs. Moreover, viruses were able to be easily removed by antibody-mediated negative selection using the cell-surface marker HN, which is expressed on Sendai virus-infected cells (Fusaki et al., 2009; Ban et al., 2011; Macarthur et al., 2012; Nishishita et al., 2012). Generation of iPSCs by polycistronic minicircle vectors have also been reported (Jia et al., 2010). PiggyBac-based transposons have been used successfully to generate iPSCs. These mobile genetic elements can be removed from a host genome by the expression of a suitable transposase, leaving no trace (Woltjen et al., 2009; Yusa et al., 2009). Whereas transposase excision leaves no residual sequences from the transgene cassettes, there are questions remaining regarding whether transposase expression can lead to nonspecific integration/excision events during transgene excision process and gross genomic rearrangements (Stadtfeld and Hochedlinger, 2009).
An alternative strategy is to avoid viral delivery by the transfection of mammalian expression plasmids while still using nucleic acid–based vectors (Okita et al., 2008). The plasmid-based reprogramming system required several rounds of transfecting two expression plasmids into the mouse embryonic fibroblasts (MEFs) but did result in iPSCs that produced teratomas and could chimerize mice without evidence of plasmid integration. Such experiments confirmed that transient expression of the core transcription factors is sufficient for reprogramming somatic cells. Production of virus-free iPSCs created a lot of hope to overcome safety concerns for potential use of iPSCs in therapeutic applications (Stadtfeld and Hochedlinger, 2010). However, the efficiency of these transient transfection reprogramming approaches was significantly lower (∼0.001%) than those achieved with integrating vectors (∼0.1–1%). This is likely due to the fact that expression of transcription factors is not maintained for a sufficient length of time to allow complete epigenetic remodeling to a pluripotent state.
To overcome the problem of the low efficiency of nonintegrating methods, a number of laboratories have used integrating vectors that have loxP sites incorporated into them flanking the reprogramming transgenes. Expression of Cre recombinase after deriving complete reprogrammed iPSCs allows for the removal of the transgenes (Kaji et al., 2009). An issue, however, is that Cre-recombinase leaves residual loxP sequences within the genome, leading to the possibility of inducing abnormal recombination (Soldner et al., 2009).
Generation of iPSCs Without Using Nucleic Acid Vectors
The use of nonnucleic acid–based transgene delivery has obvious advantages in terms of generation of integration-free iPSCs. iPSCs from human and mouse fibroblasts have been produced by introducing recombinant proteins fused to an 11R polyarginine domain (Zhou et al., 2009), whole-cell extract isolated from ESCs (Cho et al., 2010), and whole-cell extracts from genetically engineered HEK293 cells (Kim et al., 2009a). Generation of iPSCs from mouse and human fibroblasts using reprogramming factors fused to cell-penetrating peptides (CPPs) (short peptides that facilitate cellular uptake of various molecular cargos) have been described (Tang et al., 2011; Yukawa et al., 2010). Three of the most commonly used CPPs are pAntp (derived from the transcription factor Antennapedia), the herpes simplex virus VP22 protein, and the TAT protein originating from the retrovirus human immunodeficiency virus type 1 (HIV-1) (Kim et al., 2009a).
Although generation of iPSCs by delivering purified proteins is very attractive, its efficiency is very low (Kim et al., 2009a). More recently, another safer and more efficient approach for generating integration-free iPSCs has been described, which involved introduction of synthetic mRNA molecules encoding for the reprogramming factors into somatic cells (Warren et al., 2010). Using these mRNA molecules ameliorates concerns regarding insertional mutagenesis of viral genomes, and moreover its efficiency was relatively high (more than 2%, which was two orders of magnitude higher than those typically reported for virus-based derivations) (Warren et al., 2010).
An interesting strategy that has been employed in conjunction with transgene delivery is the use of small molecules that can improve or even substitute for reprogramming factors (Desponts and Ding, 2010; Nakhaei-Rad et al., 2012). These experiments have resulted in identification of molecules that can increase the reprogramming efficacy. BIX 01294, a specific inhibitor of histone methyltransferases, in conjunction with the transduction of only Oct4 and Klf4 enabled reprogramming of mouse fetal neural progenitors into iPSCs (Shi et al., 2008). Valproic acid, a histone deacetylase inhibitor, helped to increase the reprogramming efficiency of human fibroblasts with Oct4 and Sox2 more than 100-fold (Huangfu et al., 2008a,b; Li et al., 2009; Lin et al., 2009; Sun et al., 2010). Silva and colleagues combined dual inhibition of mitogen-activated protein kinase (MAPK) signaling and glycogen synthase kinase-3 (GSK3) with the self-renewal cytokine leukemia inhibitory factor (LIF) for the derivation and propagation of ground state pluripotent stem cells from mouse embryos (Silva et al., 2008). It should be noted that no single chemical compound can substitute for a reprogramming factor, and some of identified chemicals have the potential to generate iPSCs with genetic or epigenetic abnormalities in resultant cell lines, especially because many of the reported compounds are potent modulators of DNA and chromatin modifications (Yamanaka, 2009).
The Molecular Role of Factors Involved in Reprogramming
Given the significant differences between fully differentiated somatic cells and pluripotent stem cells at the epigenetic and expression levels, it may seem somewhat surprising that using only a limited number of upstream factors in this network can establish pluripotent cells from somatic cells. Among the factors used in reprogramming core are the known pluripotency factors Oct4, Sox2, and Nanog.
OCT4 encoded by Pou5f1, is a POU homeodomain transcription factor expressed in pluripotent cells of the inner cell mass (ICM), epiblast, and primordial germ cells (Boyer et al., 2005; Takeda et al., 1992). OCT4 is strongly involved in the maintenance of self-renewal of ESCs and EC cells (Matin et al., 2004). Following gastrulation, expression of Oct4 is maintained only in the germ-line lineage. Oct4−/− embryos fail to develop due to failure in ICM formation, and the embryos contain only trophectoderm cells (Avilion et al., 2003; Masui et al., 2007). Oct4 exerts its effects in a dose-dependent manner (Niwa et al., 2000). For example, Oct4 overexpression causes loss of pluripotency as cells differentiate into the primitive endoderm and mesoderm lineages (Niwa et al., 2000). On the other hand, knockdown of OCT4 expression in both human EC and ES cells gives rise to their differentiation to trophectoderm, as indicated by a marked change in morphology, growth rate, and surface antigen phenotype (Matin et al., 2004; Rassouli and Matin, 2009). This protein quantitatively controls the pluripotency by regulating the transcription of many target genes involved in development (Niwa et al., 2000). Oct4, often serves in collaboration with Sox2, which is also important for early embryo development and inhibition of differentiation (Masui et al., 2007).
Sox2 belongs to the family of high mobility group (HMG)-box transcription factors. Sox2 null embryos have defects in epiblast development and die at implantation. Sox2 also forms a complex with Oct4 and controls its own expression and downstream target genes such as FGF4 (Avilion et al., 2003; Masui et al., 2007). Sox2 is expressed in the ICM, epiblast, germ cells, and also in the primitive ectoderm and neural ectoderm (Avilion et al., 2003). The exogenous expression of Oct4 is likely to support, in cooperation with Sox2, the maintenance of the basic transcriptional framework required for ESC-like properties of iPSCs.
Nanog is expressed in the ICM and epiblast, and it is downregulated upon cell differentiation. In human and mouse ESCs, transcription of Nanog is regulated by Oct4 and Sox2. Dimerization of Nanog is crucial for its self-renewal functions (Rodda et al., 2005). ICM cells lacking Nanog differentiate into primitive endoderm, and ESCs lacking Nanog cannot sustain their pluripotency (Mitsui et al., 2003). Similar to Oct4 and Sox2, Nanog also maintains pluripotency in a concentration-dependent manner. Studies have shown that the amount of Oct4, Sox2, and Nanog is critical for the maintenance of ESCs. Important mechanisms that regulate and sustain the ESC transcriptional network are autoregulatory and feed-forward regulatory loops (Boyer et al., 2005). Therefore, it is likely that pluripotency factors such as Oct4, Sox2, and Nanog can elicit their effect by reestablishing the so called plurinet (Muller et al., 2008), thus changing the phenotype of somatic cells over time to a more pluripotent state.
Although c-Myc is a reprogramming factor, it is not explicitly associated with pluripotency. c-Myc can change the chromatin structure in somatic cells. It has been suggested that c-Myc plays an independent role early during reprogramming by induction of cellular proliferation in a similar manner to cancer cells (Mikkelsen et al., 2008; Sridharan et al., 2009). To induce its effects, c-Myc has to be expressed only during the first few days of reprogramming (Sridharan et al., 2009). Similarly, c-Myc and Klf4 premature expression in fibroblasts prior to activation of all four factors increases the efficiency and speed of reprogramming, whereas premature expression of Oct4 and Sox2 has no effect (Markoulaki et al., 2009). Moreover, c-Myc might have an effect on reprogramming later by maintaining the gene expression pattern of pluripotency, as is suggested by its presence on promoters of many genes highly expressed in ESCs. Knoepfler and colleagues showed that Myc proteins are necessary for maintaining active chromatin. Disruption of n-Myc in neuronal progenitors and other cell types results in nuclear condensation accompanied by large-scale changes in histone modifications associated with chromatin inactivation, including hypoacetylation and altered methylation (Knoepfler et al., 2006). It has been proposed that the four reprogramming factors do not act on the same set of genes, because c-Myc binds to many genes that are not bound by Oct4, Sox2, or Klf4 (Chen et al., 2008; Kim et al., 2008). Nevertheless, c-Myc shares target genes with other transcription factors, including the family member n-Myc, which can replace c-Myc in reprogramming experiments (Chen et al., 2008). Possibly, c-Myc introduced into fibroblasts causes open chromatin structure first, and then Oct4, Sox2 and Klf4 can regulate the transcription of their target genes and change the cell fate (Sridharan et al., 2009). Thus, c-Myc is likely to aid in reprogramming somatic cells by not only driving proliferation (which may be important in allowing epigenetic changes to occur) but also, at least in mouse, c-Myc is a major downstream target for LIF/STAT3 and the Wnt signaling cascades, both of which are critical for sustaining pluripotency (Kanakura et al., 2005; Yamanaka, 2007).
Klf4 belongs to the Krüppel-like family of transcription factors (Klfs). The effects of these transcription factors in cell proliferation, differentiation, and survival, especially in the context of cancer are well known (Guo et al., 2009). Klf4 can act as an oncogene or a tumor-suppressor gene (Yamanaka, 2007). Overexpression of Klf4 can maintain Oct4 expression and inhibits ESC differentiation (Li et al., 2005). Klf4 can cooperate with Oct4 and Sox2 to activate other genes in ES cells.
Lin28 is a marker of undifferentiated human ESCs (Richards et al., 2004). It encodes a cytoplasmic mRNA-binding protein that binds to and enhances the translation of the insulin-like growth factor-2 (IGF-2) mRNA (Balzer and Moss, 2007; Polesskaya et al., 2007). Lin28 expression is downregulated during the differentiation of mouse and human ESCs (Shuang and EnKui, 2008).
Pluripotency factors suppress the differentiation-associated genes with recruitment of suppressive chromatin remodeling complexes such as polycomb (Boyer et al., 2006; Lee et al., 2009), resulting in histone deacethylation and H3K27 trimethylation. The binding of pluripotency factors to their target genes might be facilitated by nucleosome remodelers such as Chd1 (Gaspar-Maia et al., 2009) and BAF (Singhal et al., 2010), both of which increase reprogramming efficiencies and kinetics when overexpressed.
Although there has been some success in improving delivery methods and reprogramming efficiencies, it is still far from an optimal method. Genome-wide expression analyses and global histone modifications have confirmed that iPSCs and ESCs are highly similar (Maherali et al., 2007; Okita et al., 2007; Wernig et al., 2007). On the other hand, important differences between the two cell types have also been reported. For instance, iPS cell-derived neural cells have shown increased tumor formation after transplantation into the brain (Miura et al., 2009). In addition, human iPS cell-derived early blood progenitor cells appear to undergo premature senescence (Feng et al., 2010). Furthermore, some differences in DNA methylation (Deng et al., 2009; Pick et al., 2009) and mRNA and miRNA (Wilson et al., 2009) expression, and also at the proteome level (Faradonbeh et al., 2012), have been reported between ESCs and iPSCs. It is likely that much more careful control of the stoichiometry of different reprogramming factors and/or concentration of small molecules will be required to improve derivation efficiencies and produce fully reprogrammed iPSCs. Detailed analysis of the gene expression and epigenetic changes will need to be mapped as cells are reprogrammed to establish how a given reprogramming strategy traverses through the different states that lie between a fully differentiated somatic cell and a bone fide pluripotent cell. The requirement for detailed time-course information on the reprogramming process points to the need for much more efficient reprogramming strategies and increased use of reporters of potential markers of the intermediate states and single cell–based analyses.
Applications
As shown in Figure 1, iPSCs are exciting research tools that also have therapeutic potential for both disease modeling and cell therapy (van der Heyden and Jonsson, 2012). Using these cells as models will accelerate the exploration of mechanisms that control pluripotency (Chun et al., 2010). A second major area of interest is cellular therapy and, in particular, the use of patient-matched cells for transplantation. Several papers have reported the use of iPSCs in cell animal-based therapy models. Dopaminergic neurons derived from reprogrammed fibroblasts were shown to functionally integrate into the donor brain and improve symptoms of rats with Parkinson's disease (Wernig et al., 2008). In another study, phenotypic correction of hemophilia A could be achieved in mice transplanted with heterologous iPSC-derived endothelial progenitor cells (Xu et al., 2009). One of the important objectives in the use of iPSCs in personalized medicine is the differentiation of these cells to functional target cells. In a study, iPSCs were differentiated successfully to functional neurons, expressing important dopaminergic and midbrain markers such as βIII-tubulin, tyrosine hydroxylase (TH), aromatic
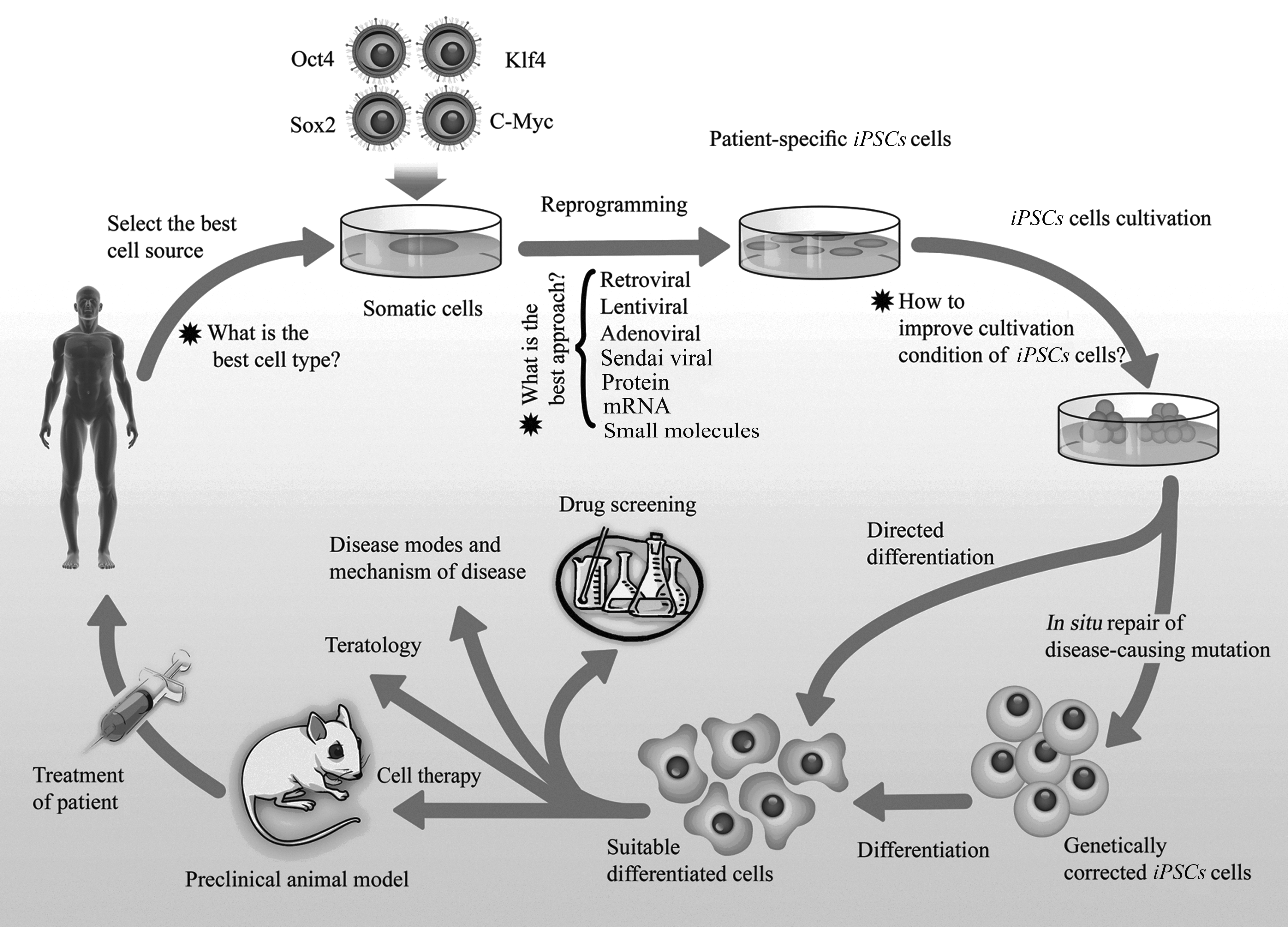
iPSCs as a powerful and promising tool for therapy and research.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by degeneration of motor neurons. iPSCs derived from a woman with familial ALS were guided to differentiate into motor neurons. Transplantation of these motor neurons might finally be used to treat ALS in the future (Dimos et al., 2008). Although cell therapy may be an attractive choice for treatment of degenerative diseases such as Alzheimer's disease (Abdel-Salam, 2011), type 1 diabetes (Neshati et al., 2010), and spinal cord injury (Wang et al., 2012), there are some caveats in using iPSCs. Undoubtedly the iPSC method of reprogramming adult cells, without using donated oocytes or embryos, would obviously be technically and ethically more advantageous. The most obvious advantage of this technology is generation of patient-specific stem cells to overcome the problem of immune rejection (Boland et al., 2009; Chun et al., 2010; Park et al., 2008). However, a recent study has reported the immunogenicity of iPSCs after direct transplantation of undifferentiated cells into syngenic mice. They reported that even in a syngenic mouse, the transplantation of immature iPSCs induced a T cell–dependent immune response. They used three types of experimental models: an iPSC autograft, an ESC autograft, and an ESC allograft (Zhao et al., 2011). It is important to note the results obtained using undifferentiated iPSCs might simply reflect immunoreaction against undifferentiated pluripotent cells. In addition, the authors compared only one line of syngenic ESCs with several lines of iPSCs (Okita et al., 2011).
In another study in 2007, Jaenisch's group generated iPSCs from tail-tip fibroblasts of humanized sickle cell anemic mice. Then, a sickle cell gene mutation in these cells was corrected by homologous recombination, and the cells were directed to differentiate into hematopoietic progenitor cells. Finally, the corrected iPSCs were transplanted back into donor mice and successfully treated sickle cell anemia without immune rejection, thus demonstrating the therapeutic potential of iPSCs (Hanna et al., 2007). However, there still remains the issue of whether it is realistic to think of being able to generate potentially thousands of different patient-specific iPSCs, given the timescale of derivation, differentiation to the transplantable cell type, and, above all, the costs required to meet current regulatory requirements for transplantable cell products.
Another important application of iPSCs is generation of disease-specific models. So far, many disease-specific iPSCs have been generated (Dimos et al., 2008; Ebert et al., 2009; Hanna et al., 2007; Lee et al., 2009; Ma et al., 2012; Raya et al., 2009; Yi et al., 2012). In 2009, the first iPSC line containing a genetic disorder, a human sickle cell anemia iPSC line, was successfully established (Ye et al., 2009). The establishment of a disease iPSC bank would help to identify mechanisms for specific diseases and to discover effective treatments for them (Park et al., 2008; Zaehres and Scholer, 2007). In principle, this method can be used for treating all diseases that are caused by a known mutation and can be treated by cell transplantation.
Areas of Future Research
iPSC technology is still a relatively new area, and therefore much detailed research will need to be carried out if induced pluripotency is to be used in regenerative medicine. Currently, the majority of iPSC generation approaches are still using virus-based integrating systems. These methods have limitations, such as low efficiency or insertional mutagenesis (Okita et al., 2007). Furthermore, some of the factors that are currently used, such as c-Myc and Klf4, are oncogenes and their reactivation can cause tumorigenesis (Sun et al., 2010). It has also been shown that iPSCs keep an epigenetic memory of their original tissue, which may cause restrictions in their differentiation capacity (Kim et al., 2010). Much more research is required to uncover the mechanisms underlying factor-induced reprogramming. Fundamental questions still remain unanswered. For example, what is the exact role of every single introduced factor? How are these factors working together? Do recognized sets of reprogramming factors truly represent a core regulatory circuit? In what order do these factors work? What is the appropriate level of expression of each factor that can trigger somatic cell nuclear reprogramming? What are other unknown factors? Which factors have more safety and better performance? Which factors can be replaced by other agents, and what are those other agents? In a reprogramming experiment, multiple iPSC lines were usually generated that are not identical. So, establishing a reliable standard protocol for identifying proper reprogrammed iPSCs is necessary. Studies using inducible promoters or nonintegrating methods should allow for the elucidation of optimal stoichiometry for reprogramming. If we can control the timing and the levels of expression of these genes, the mechanisms involved in induced reprogramming can be studied more carefully and thoroughly. Another question is exactly which cells are able to be reprogrammed? iPSCs as disease models can move the cancer field forward by allowing more types of experiments to be performed. Use of patient-specific fibroblasts as a feeder layer may help to overcome this problem (Richards et al., 2002; Unger et al., 2008).
Although not unique to iPSCs, it is important to note that molecular and morphological characteristics of stem cells may change upon long-term culture as they undergo progressive adaptation to in vitro culture (Enver et al., 2005; Matsui et al., 1992). In the case of iPSCs, similar changes to ESCs have been identified as appearing over long-term culture (Laurent et al., 2011), and the reprogramming process itself may induce mutation (Hussein et al., 2011). It is far from clear how these epigenetic changes affect tumorigenicity, differentiation capacity, and function of cells derived from iPSCs. The eventual use of iPSCs in clinical applications is intimately tied up with progress in controlling the epigenetic and genetic changes in pluripotent cells as they are expanded. Ultimately, though, any clinically related uses for iPSCs are going to rely on significant progress being made in the ability to control the differentiation from pluripotent cells to mature, functional cell types.
Footnotes
Acknowledgment
We are grateful to Dr. Paul J. Gokhale from University of Sheffield for his great input and proofreading the manuscript.
Author Disclosure Statement
No competing financial interests exist.