
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) represent a novel cell source for regenerative therapies. Many emerging iPSC-based therapeutic concepts will require preclinical evaluation in suitable large animal models. Among the large animal species frequently used in preclinical efficacy and safety studies, macaques show the highest similarities to humans at physiological, cellular, and molecular levels. We have generated iPSCs from cynomolgus monkeys (Macaca fascicularis) as a segue to regenerative therapy model development in this species. Because typical human immunodeficiency virus type 1 (HIV-1)-based lentiviral vectors show poor transduction of simian cells, a simian immunodeficiency virus (SIV)-based vector was chosen for efficient transduction of cynomolgus skin fibroblasts. A corresponding polycistronic vector with codon-optimized reprogramming factors was constructed for reprogramming. Growth characteristics as well as cell and colony morphology of the resulting cynomolgus iPSCs (cyiPSCs) were demonstrated to be almost identical to cynomolgus embryonic stem cells (cyESCs), and cyiPSCs expressed typical pluripotency markers including OCT4, SOX2, and NANOG. Furthermore, differentiation in vivo and in vitro into derivatives of all three germ layers, as well as generation of functional cardiomyocytes, could be demonstrated. Finally, a highly efficient technique for generation of transgenic cyiPSC clones with stable reporter expression in undifferentiated cells as well as differentiated transgenic cyiPSC progeny was developed to enable cell tracking in recipient animals. In conclusion, our data indicate that cyiPSCs represent a valuable cell source for establishment of macaque-based allogeneic and autologous preclinical cell transplantation models for various fields of regenerative medicine.
Introduction
Considering the substantial physiological differences among species (Gandolfi et al., 2011; Wessels and Sedmera, 2003), other hurdles will require exploration in large animal species that are more similar to humans than mice and rats (Gandolfi et al., 2011). For example, due to major differences on molecular, morphological, and functional levels, results from murine cardiovascular disease models frequently fail to translate to the human system under investigation. For instance, mice and humans exhibit different sizes of myocytes, different myocardial capillary densities (Rakusan and Nagai, 1994; Stoker et al., 1982), and differing proportions of β-myosin versus α-myosin subunits within the ventricle (Doevendans et al., 1998; Swynghedauw, 1986). Moreover, human cardiomyocytes appear to be unable to functionally couple to mouse myocardium due to highly differing contraction rates (van Laake et al., 2008). Functional effects of cellular cardiomyoplasty have to be demonstrated in large animals, if possible in a syngeneic or allogeneic transplant setting. Similarly, the risk of teratoma and tumor formation after application of enriched iPSC derivatives cannot be addressed adequately in mice or in xenogeneic transplant models. When using immunoincompetent [e.g., severe combined immunodeficiency (SCID)] mice, there might be overestimation of tumor risk due to absence of T cells and functional natural killer (NK) cells that are responsible for tumor cell elimination. In contrast, in xenogeneic transplantation models, when transplanting human cells into pigs, tumor risk might be underestimated due to xenogeneic immunoreactions.
Recently, iPSCs from pigs (Esteban et al., 2009; Ezashi et al., 2009) and dogs (Lee et al., 2011; Luo et al., 2011; Shimada et al., 2011) have been described. However, porcine iPSCs appear to be much different from human iPSCs (hiPSCs), and establishment of optimal culture conditions, efficient genetic manipulation, or specific differentiation into certain lineages are ongoing research efforts and may not be expected to yield results in the very near future.
This is in contrast to nonhuman primate pluripotent stem cells (PSCs), especially from Old World monkeys, and, unlike porcine embryonic stem cells (ESCs), ESCs from macaques show a high degree of similarity to human cells (Olmer et al., 2010; Schwanke et al., 2006b). Due to the importance of macaques as validated large animal models of human physiology and disease, iPSCs have been generated from Macaca mulatta (rhesus monkey) (Chan et al., 2010; Liu et al., 2008; Zhu et al., 2011), the most frequent macaque species used in experimental research, and from M. nemestrina (pigtailed macaques) (Zhong et al., 2011). iPSCs have also been generated from M. fascicularis (cynomolgus monkey) (Deleidi et al., 2011; Okahara-Narita et al., 2012; Okamoto and Takahashi, 2011), the most frequent primate model used in safety pharmacology (Authier et al., 2007; Champeroux et al., 2009), a discipline that is critical for preclinical evaluation of stem cell–based therapies (Bjugstad et al., 2008; Deleidi et al., 2011; Emborg et al., 2008; Redmond et al., 2007; Roitberg et al., 2006). Notably, all macaque iPSC lines described so far have been generated through transduction with γ-retroviral vectors expressing individual key reprogramming factors. Conventional human immunodeficiency virus type 1 (HIV-1)-derived lentiviral vectors commonly used for reprogramming of human cells (Haase et al., 2009; Yu et al., 2007) are not applicable in simian cells due to a specific intracellular block of HIV infection (Perron et al., 2007; Sakuma et al., 2007; Stremlau et al., 2006). In contrast, the related simian immunodeficiency virus (SIV) is not blocked, and SIV-based vectors represent an efficient alternative to HIV-1 vectors for genetic manipulation of simian cells (Mangeot et al., 2000; Negre et al., 2000).
To improve the reprogramming of macaque cells, we have engineered a straightforward “all-in-one” SIV-based lentiviral vector (Mangeot et al., 2002) co-expressing codon-optimized reprogramming factors (Warlich et al., 2011). Using this vector, cynomolgus monkey iPSCs were generated that could be efficiently differentiated into functional cardiomyocytes, which were characterized on a molecular, electrophysiological, and pharmacological level. Finally, reporter cynomolgus (cy) iPSCs clones with stable long-term enhanced green fluorescent protein (eGFP) expression in undifferentiated cells, and their differentiated derivatives were generated to enable tracking of donor cells as a prerequisite for development and validation of preclinical macaque models of regenerative cell-based therapies.
Materials and Methods
Preparation of lentiviral vector stocks and assessment of transduction efficiencies
For determination of transduction efficiencies, the SIV-based self-inactivating lentiviral vector plSh S EF1α hrGFP WPRE and the HIV-1-based lentiviral vector plSh H EF1α hrGFP WPRE were used. The vector plSh S is a combination of the transfer vector pSIVRMESGAE (kindly provided by Dr. Didier Nègre, École Normale Supérieure de Lyon, France) and the established shuttle system that has been described previously (Gruh et al., 2005). The vector plSh H belongs to the shuttle vector system described in Gruh et al. (2005).
For reprogramming an expression cassette containing the four codon-optimized reprogramming factors OCT4, KLF4, SOX2, and c-MYC (Warlich et al., 2011) under the control of the spleen focus-forming virus (SFFV) promoter, all coupled through 2A sites, together with the reporter gene dTomato red, coupled through an internal ribosome entry site (IRES), was cloned into the SIV-based vector plSh S. For virus production we co-transfected the transfer vector plSh S SF. hOct34. hKlf4. hSox2. hMyc. i2dTomato, plSh S EF1α hrGFP WPRE or plSh H EF1α hrGFP WPRE, the packaging plasmid pSIV3+ for SIV-based vectors, and psPAX2 for HIV-1-based vectors and the envelope-coding plasmid pMD2.G into HEK293T cells using the calcium phosphate DNA precipitation method (Naldini, 1998). The supernatant was collected after 48 h and concentrated by 16 h centrifugation at 18,000×g. The resulting virus pellet was resuspended in Dulbecco's modified Eagle medium (DMEM) and stored until use at −80°C (Wunderlich et al., 2008).
Vector titration and determination of integrated vector copies by means of quantitative real-time PCR
The active biological titer of our vector preparations after the final concentration step was determined by quantitative real-time PCR. HeLa229 cells were transduced with the vectors. After three passages, HeLa229 cells were washed twice with phosphate-buffered saline (PBS). Detached cells (1×106) were lysed in 100 μL of proteinase K (200 μg/mL in PCR buffer) for 3 h at 56°C followed by a 10-min inactivation at 95°C. These crude extracts were diluted 1:10, and 5 μL served as a template for the PCR reactions. To detect viral long terminal repeat (LTR) sequences within the genomic DNA of the target cells (genome titer), forward primer 5′-AGCTTGCCTTGAGTGCTTCA- 3′, reverse primer 5′-TGACTAAAAGGGTCTGAGGGA-3′, and probe 5′-FAM TGCCCGTCTGTTGTGTGACTCTG-TAMRA-3′ were used. Amplification reactions were performed with HotGoldStar polymerase (qPCR Core kit No ROX; Eurogentec, Seraing, Belgium) according to the manufacturer's instructions, using an iCycler iQ real-time PCR detection system (Bio-Rad Laboratories, Munich, Germany). The copy number of proviral DNA was measured by comparison with a plasmid standard curve. PCR data were expressed as mean copy numbers based on triplicate PCR samples of two independent transduction experiments for titration of each vector. For assessment of integrated vector copies in target cells, three independent transduction experiments were analyzed with the same method.
Transduction efficiencies on cynomolgus skin fibroblasts were determined after transduction of adherent cynomolgus fibroblasts with a multiplicity of infection (MOI) (Emborg et al. 2008) of 1 for 4 h. Then 2 mL of fresh culture medium was added, and the cells were cultivated overnight prior to complete medium change. In the case of GFP-expressing cells, the transduced cells were measured by flow cytometry after the second-passage posttransduction.
Isolation and culture of fibroblasts and induction of PSCs
Skin biopsies from cynomolgus monkeys (all 5 years of age) were obtained from Covance (Münster, Germany). Skin fibroblasts were isolated according to standard protocols and cultivated in fibroblast culture medium [DMEM supplemented with 1 mM
One day prior to transduction, the cynomolgus fibroblasts (2×105) were seeded onto six-well plates (Thermo Scientific, Waltham, MA, USA). The adherent cells were transduced with the concentrated lentiviral vector plSh S SF.hOct34.hKlf4.hSox2.hMyc.i2dTomato (MOI 10) in fibroblast culture medium containing 8 μg/mL Polybrene (Sigma-Aldrich, Deisenhofen, Germany). The transduced fibroblasts were then cultured for 6 days in fibroblast culture medium without splitting. On day 6, the transduced fibroblasts were trypsinized and seeded on top of irradiated murine embryonic fibroblasts (MEFs). On day 7, the culture medium was changed to human ESC medium [Knockout DMEM supplemented with 20% Knockout Serum Replacement (KSR), 1 mM
Culture of undifferentiated cynomolgus iPSCs and ESCs
ESCs and iPSCs were cultivated and expanded under standard ESC culture conditions in knockout DMEM supplemented with 20% KSR, 1 mM
Karyotype analyses
After treatment of adherent iPSCs for 30 min with colcemide (Invitrogen) at final concentrations of 0.025, 0.05, and 0.1 μg/mL, cells were dissociated with 0.83% trypsin EDTA (Invitrogen). After hypotonic treatment with 60 mM or 75 mM potassium chloride for 20 min at 37°C and cytogenetic cell preparation, fluorescence R-banding was performed according to standard procedures.
Differentiation of cynomolgus ESCs and iPSCs in vitro and in vivo
For embryoid body (EB)–based differentiation in vitro, cynomolgus ESCs/iPSCs were dispersed into small clumps, transferred into six-well, low-attachment plates (Greiner, Frickenhausen, Germany), and cultured for 1 day in SF medium (knockout medium and Ham's F12 (1:1) supplemented with 0.05% N2 stock solution (100×), 0.1% B27 stock solution [50×), 1% nonessential amino acid stock, 1 mM
Teratomas were induced by injection of a cell Matrigel mixture (1:1) (BD, Heidelberg, Germany) into a hind limb muscle of SCID-beige mice. After 8–10 weeks, the animals were sacrificed, and teratomas were examined by an experienced pathologist according to Wesselschmidt and Schwartz (2011).
Analysis of mRNA expression by reverse transcription PCR and quantitative real-time PCR
Total RNA was prepared using the RNeasy Kit (Macherey-Nagel, Dueren, Germany) and reverse transcribed with Superscript II (Invitrogen) using oligo(dT) primers according to the manufacturer's instructions. For reverse transcription PCR (RT-PCR), 1 μL of cDNA was amplified by PCR with GoTaq polymerase (Promega, Madison, WI, USA). Sequences of primers are shown in Table S1 (see Supplementary Data at www.liebertpub/cell/). Quantitative real-time PCR was performed in triplicate using a Mastercycler® ep realplex2 (Eppendorf, Hamburg, Germany) and the Absolute™ QPCR SYBR® Green Mix (ABgene, Epsom, Surrey, UK). The size of amplicons and the absence of nonspecific products were controlled by melting curves. Sequences of primers are shown in Table S2. Starting quantities of target cDNAs were calculated by comparing threshold cycle (CT) values of each sample to CT values of the respective standard curve using Mastercycler® ep realplex Software Version 2.0 (Eppendorf). Expression levels of target genes were normalized to β-actin transcript levels. Data are expressed as the mean±standard error of the mean (SEM) of normalized gene expression levels from three experiments. Notably, mRNA expression levels of endogenous OCT4 and transgenic OCT4 (relative to β-actin) are directly comparable, because these levels were not calculated through the ΔCT approach, but through co-measured dilution series (for endogenous OCT4, transgenic OCT4, and β-actin).
Immunocytological staining and assessment of the efficiency of cardiac differentiation
Immunostaining of undifferentiated iPSCs and their differentiated derivatives was performed after fixation with 4% paraformaldehyde and subsequent blocking with 5% donkey/goat serum and 0.25% Triton X-100 (Sigma-Aldrich) diluted in Tris-buffered saline for 20 min at room temperature.
Primary antibodies that were used are described in Table S3. The plates were incubated for 1 h at room temperature with the primary antibody diluted in phosphate-buffered saline (PBS) (without Ca2+/Mg2+) with 1% bovine serum albumin (BSA), then rinsed three times with PBS. Further incubation was performed with the appropriate secondary antibodies (DyLight 488, 549, 649; Jackson ImmunoResearch, Suffolk, UK) for 30 min at room temperature. The plates were rinsed once more, counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich), and analyzed with an Axio Observer A1 microscope (Carl Zeiss MicroImaging, Goettingen, Germany). Contracting versus noncontracting EBs were counted on day 27 of differentiation.
Microelectrode mapping
Extracellular recordings of field potentials (FPs) were performed using a multielectrode array (MEA) data acquisition system (Multi Channel Systems, Reutlingen, Germany) (Mauritz et al., 2008). For this analysis, beating areas in plated cyiPS-EBs were removed mechanically and plated onto MEA culture plates coated with gelatin and fibronectin (BD, Heidelberg, Germany). For recording of extracellular FPs from the visibly beating syncytia, the MEA dish was transferred into a temperature-controlled MEA1060UP-USB amplifier. Recordings were performed for 1 min per condition after 10 min of incubation for the baseline and also after 10 min after adding the different substances. The amplifier together with the MEA was put into an incubator (CO2 incubator MCO-10AIC, Sanyo, Munich, Germany) for the whole time of recording to stabilize the surrounding conditions. The recorded data were used to analyze the field potential duration (FPD). The FPD was shown to be in accordance to the action potential duration (Banach et al., 2003; Halbach et al., 2003; Omura, 1970). The frequency of contractions was calculated by measuring the distance between respective FP minima and denoted interspike interval. An important characteristic of cardiomyocyte physiology is the responsiveness to neurohormonal regulation. To prove these characteristics, we looked for the effect of isoproterenol hydrochloride on the beating frequency of the cardiomyocytes. Additionally, we added different drugs and studied the effects of the exogenous drugs on the FPD and the beating frequency. The drugs were diluted in ultrapure water and added to 1 mL of cell culture medium. We used verapamil and quinidine (both Sigma-Aldrich) at a concentration of 100 nM (verapamil) and 30 μM (quinidine).
Cell transfection and establishment of transgenic clones
Transgenic cell clones were generated through introduction of the reporter gene construct pCAGGS2 containing green fluorescent protein (GFP) under the control of the chicken β-actin (CBA) promoter combined with the human cytomegalovirus immediate early (CMV-IE) enhancer (together termed CAG promoter). Before electroporation, pCAGGS2 was linearized using BsmI (New England Biolabs, Frankfurt, Germany).
All transfections were performed using the Neon™ transfection system (Invitrogen). For harvesting, the cells were treated using collagenase IV (Invitrogen), followed by an additional incubation step with TrypLE™ (Invitrogen) for single-cell dissociation according to the manufacturer's specifications. For transfection, 1.5×106 of undifferentiated detached cyiPSCs were washed once with PBS (without Ca2+/Mg2+) and pelleted at 1000 rpm for 5 min, resuspended in 110 μL of resuspension buffer R, and transferred with 40 μg of pCAGGS2 plasmid DNA into sterile microcentrifuge tubes. The cells were electroporated with two pulses at 1200 V for 20 msec and plated onto Matrigel-coated 6-cm dishes in preincubated conditioned medium supplemented with 10 μM ROCK-Inhibitor Y-27632 (supplied by the Institute for Organic Chemistry, Leibniz University Hannover) (Palecek et al., 2011). At 9 days after electroporation, the cells were subjected to fluorescence-activated cell sorting to obtain transgene-expressing cells. Approximately 4×104 GFPpos cells were reseeded at low density into Matrigel-coated 6-cm dishes. Transgene-expressing colonies were manually selected on day 20 after electroporation, transferred to irradiated feeder-layer based cultures, and expanded clonally. For further analysis, the clones were differentiated to verify a stable transgene expression during differentiation.
Characterization of transgenic clones
Established clones were analyzed for the expression of typical pluripotency-associated markers via immunfluorescence staining. Stable expression of fluorescence reporter genes in undifferentiated cells as well as differentiated progeny was monitored by microscopy.
Statistical analysis
Results are reported as mean±SEM. Analysis of variance (ANOVA) and Bonferroni statistical analyses were performed. Values with p<0.05 were considered to be statistically significant.
Results
Transduction of cynomolgus fibroblasts with HIV-1- or SIV-derived lentiviral vectors
As demonstrated recently, HIV-1, although able to enter macaque target cells, is not able to establish an infection in Old World monkey cells (Besnier et al., 2002; Cowan et al., 2002; Munk et al., 2002; Owens et al., 2003; Rahm et al., 2011), implying that HIV-1-derived vectors are unsuitable for efficient gene transfer into such cells. SIV-derived vectors have been shown to represent a good alternative to overcome this problem (Mangeot et al., 2002; Mangeot et al., 2000; Negre et al., 2000).
We decided to use a second-generation, self-inactivating lentiviral vector system based on SIVmac (Mangeot et al., 2002) with safety features comparable to common HIV-1-based vectors. In direct comparison with an HIV-1-based vector system, a SIVmac-based vector was evaluated for efficiency in transducing cynomolgus monkey fibroblasts. We transduced the adherent cells with vectors containing hrGFP under the control of the CMV promoter with a MOI of 1. Two passages after transduction, the fibroblasts transduced with the HIV-1-based vector showed almost no GFP-positive cells, whereas the fibroblasts transduced with the SIV-based vectors showed almost 60% GFP-positive cells (Fig. 1A). On the basis of this result, a SIV-based polycistronic vector expressing codon-optimized reprogramming factors was constructed (Fig. 1B) and used for induction of PSCs from cynomolgus fibroblasts.
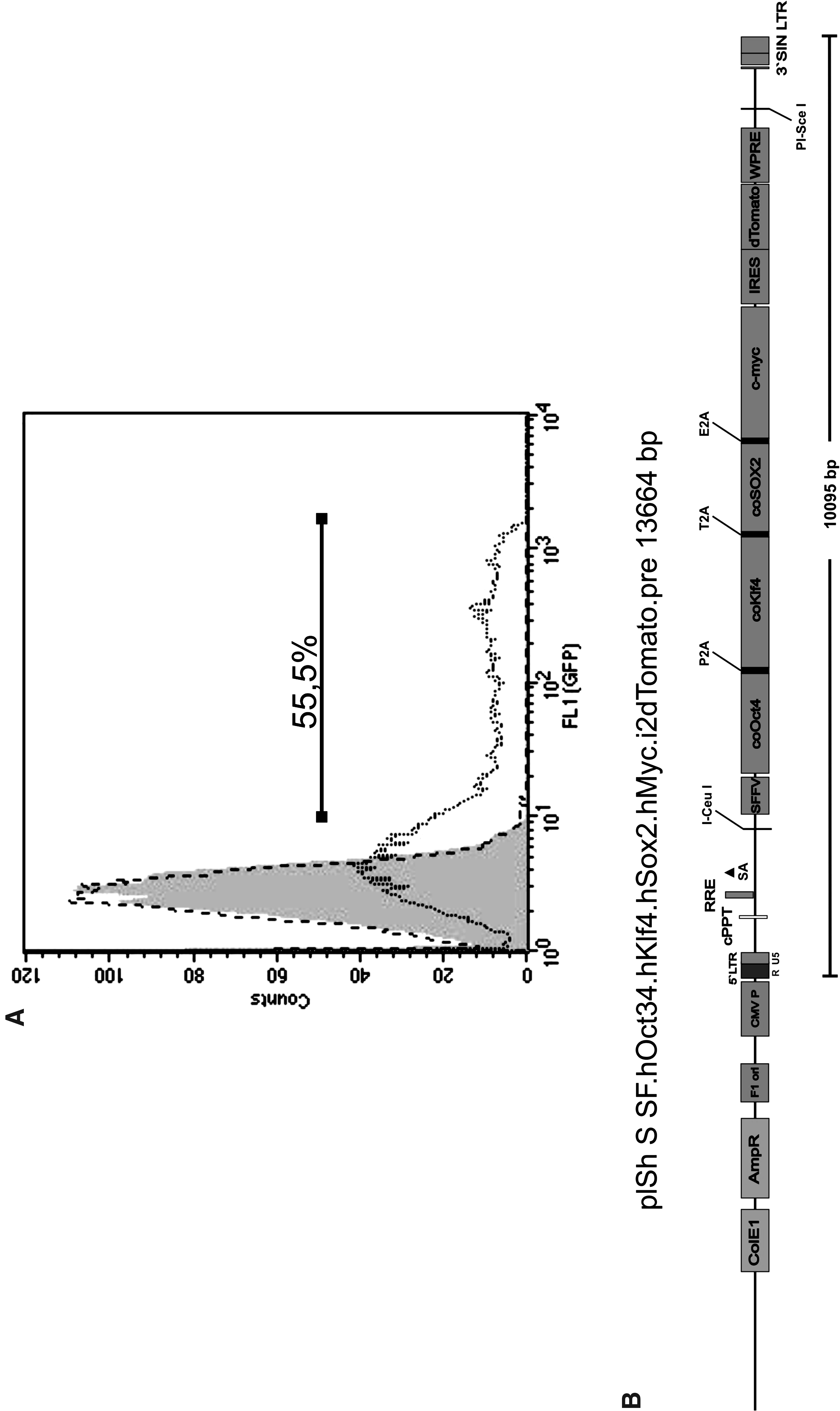
Efficient transduction of cynomolgus skin fibroblasts with SIVmac-based lentiviral vectors as compared to HIV-based vectors. (
Induction of pluripotency in M. fascicularis skin fibroblasts
To facilitate reprogramming vector construction and reprogramming itself, we took advantage of a previously developed multicistronic expression cassette for simultaneous tightly adjusted expression of reprogramming factors (Warlich et al., 2011). In this expression cassette, codon-optimized OCT4, SOX2, KLF4, and c-MYC, coupled with three 2A protease sites, were placed under control of the SFFV promoter, which mediates efficient expression in fibroblasts and other somatic cell types but is rapidly silenced in cells undergoing epigenetic remodeling (Warlich et al., 2011). After integrating this optimized expression cassette into the SIVmac vector system mentioned above, we used the resulting reprogramming vector plSh S SF.hOct34.hKlf4.hSox2.hMyc.i2dTomato (Fig. 1B) to reprogram cynomolgus fibroblasts applying a MOI of 10. Initially, the colonies forming showed a compact, multilayered, three-dimensional appearance. After the second passage, the colony morphology changed and the colonies adopted the typical flat and more single-layered appearance similar to established cyESC lines (Fig. 2A). Notably, the morphology of cyESCs as well as cyiPSCs much more closely resembles human pluripotent cell lines than murine pluripotent cells. The calculated efficiency of iPSC generation was ∼0.003%.

CyiPSCs show typical ESC morphology and express typical ESC markers including endogenous pluripotency factors. (
CyiPSC clones show typical characteristics of ESCs
CyiPS could be cultured under typical hESC conditions (Thomson et al., 1998). We have cultured the cyiPS clones for up to 40 passages using this methodology. Karyotype analyses were performed at passage 32 and did not detect any abnormalities (Fig. S1). Immunocytological staining of cyiPS cells revealed expression of pluripotency markers, including OCT4, SOX2, NANOG, Tra-1-60, Tra-1-81, and SSEA-4 (Fig. 2B). In contrast, no SSEA-3 expression was detected (Fig. 2B).
In agreement with recently published data (Warlich et al., 2011) on a very similar polycistronic reprogramming vector based on a HIV-1 vector backbone, no expression of transgenic dTomato could be observed in our cyiPSC clones (data not shown).
Applying highly sensitive quantitative RT-PCR, some OCT4 transgene expression was still detectable but appeared to be much lower than expression of endogenous OCT4 in all clones (Fig. 2C). Furthermore, we observed a varying degree of silencing between different clones. CyiPS3 exhibited the highest expression of transgenic codon-optimized OCT4, whereas cyiPS2 exhibited 50-fold lower expression, the lowest transgene expression level observed in any clone. The level of the endogenous expression of OCT4, NANOG, and SOX2 was on similar levels compared to the cyESC line MF12 (Fig. 2C, left panel) and was considerably higher than in the source fibroblasts, thus indicating full reprogramming of cyiPSCs. Importantly, the level of remaining transgenic OCT4 in the reprogrammed cyiPSC clones was very low compared to the endogenous OCT4 expression (Fig. 2C, right panel).
CyiPSCs differentiate into derivatives of all three germ layers in vitro and in vivo
In vitro differentiation of cyiPSCs and cyESCs in EBs (Liebsch et al., 2011) was performed to assess the differentiation capacity of cyiPSCs. In general, expression of differentiation markers of all three germ layers increased during differentiation comparable to the cyESCs (Fig. 3). Although the ectodermal markers NESTIN, SPARC, and β3-Tubulin could already be detected in cyESC and cyiPSC expansion cultures, our results indicate an upregulation during differentiation in the case of WNT1 and FGF5, and TTR markers of endoderm. They were upregulated between days 4 and 17 of differentiation. A strong transient increase of expression of the mesendodermal marker Brachyury (T) was observed from days 2 to 7 of culture.
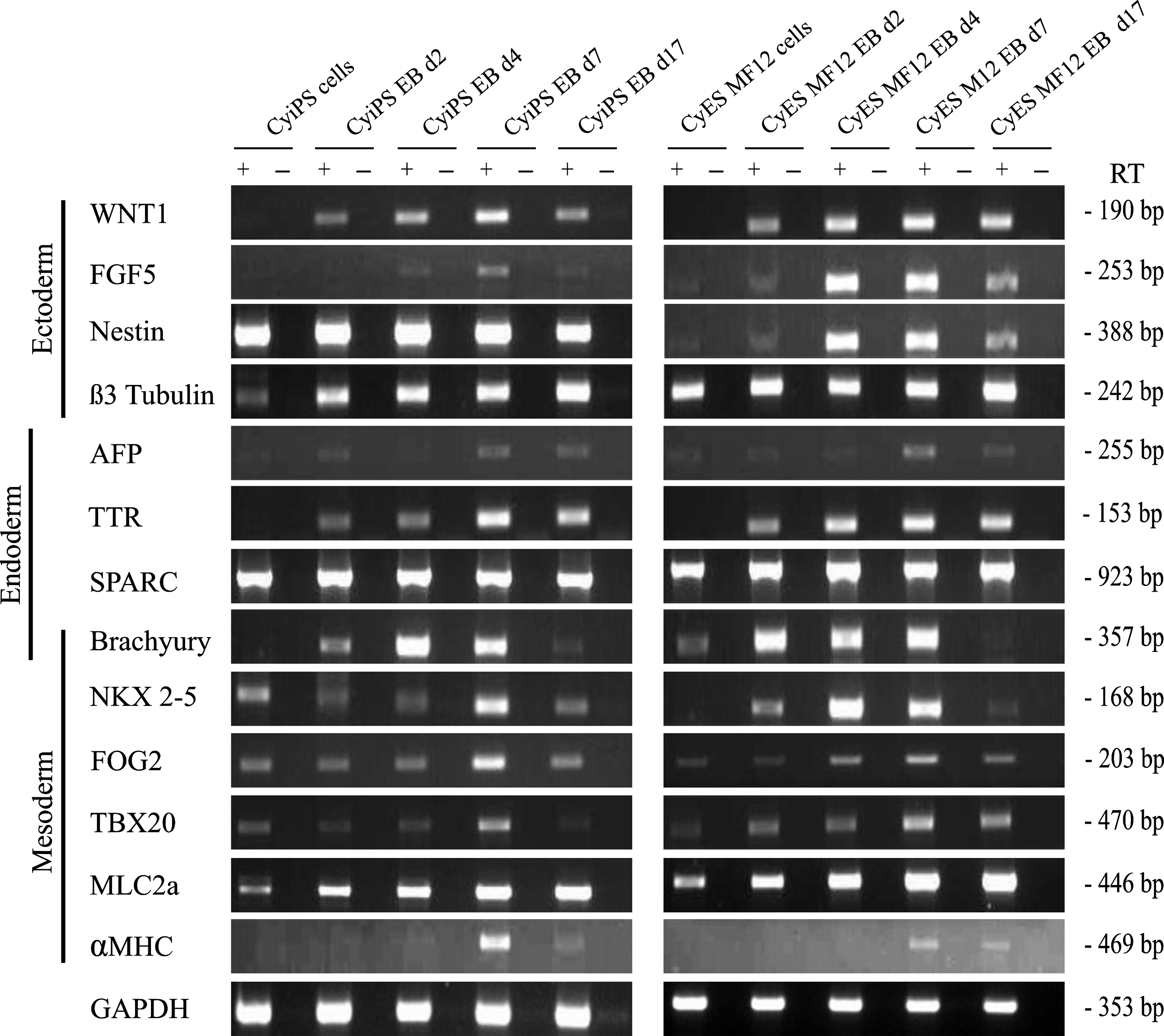
Differentiated derivatives of cyiPSCs express ectodermal, endodermal, and mesodermal marker proteins, as shown by RT-PCR. EB-based differentiation of cyiPS2 cells induced mRNA expression of markers for mesoderm and endoderm, cardiac mesoderm, and CMs, with an expression pattern comparable to cyESCs. Semiquantitative RT-PCR was performed on undifferentiated cells and cells at different time points after induction of differentiation. To exclude false-positive results due to contamination with genomic DNA, adequate controls without RT (−) were used.
RT-PCR results were confirmed by immunocytology for β3-tubulin (ectoderm), alpha-actinin (mesoderm), and AFP and SOX17 (endoderm) (Fig. 4A). Transplantation of undifferentiated cyiPS cells into immunodeficient SCID-beige mice resulted in the formation of well-differentiated teratomas containing derivatives of all three germ layers (Fig. 4B, Figs. S2 and S3).
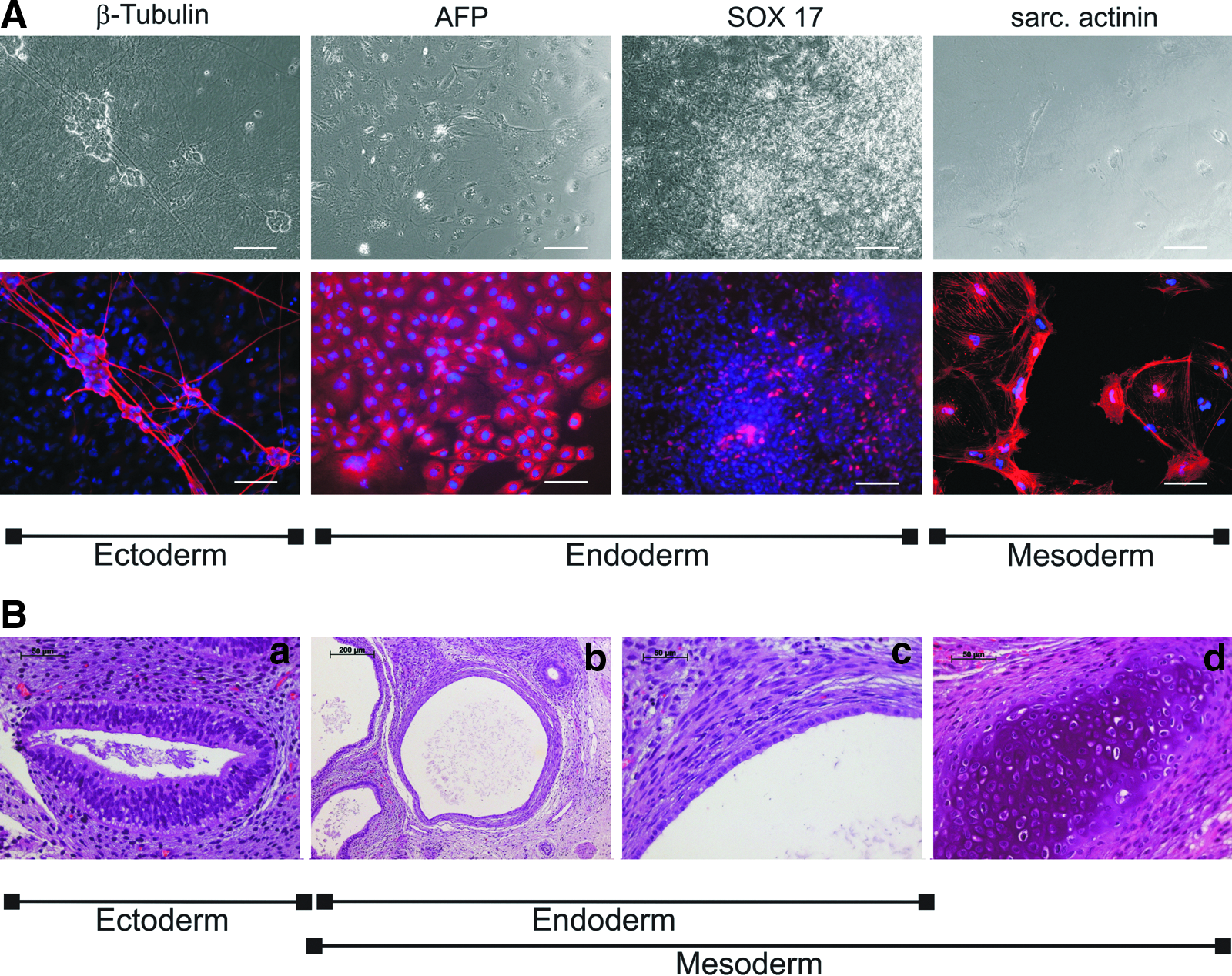
CyiPS cells differentiate into ectodermal, endodermal, and mesodermal derivatives both in vitro and in vivo. (
Differentiation of cyiPSCs into spontaneously contracting functional cardiomyocytes
All three cyiPSC clones differentiated into spontaneously contracting myocytes. We observed on average in all three cyiPS clones up to 12.8% spontaneously contracting EBs (Fig. 5). A more detailed analysis was performed for differentiation of the cyiPS2 into cardiomyocytes as described below.
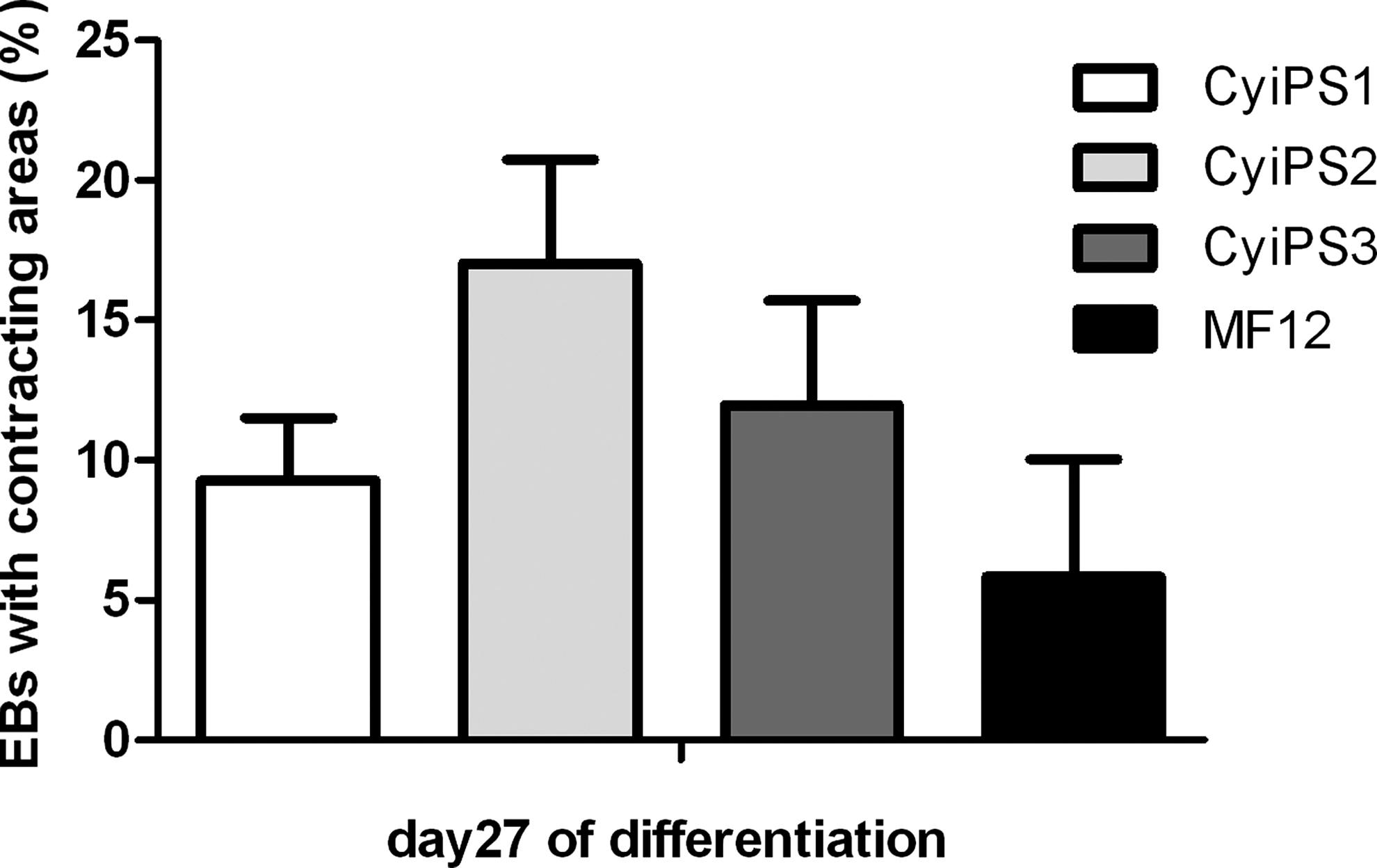
Cardiac differentiation of cyiPSCs. The numbers of EBs with contracting areas were counted on day 27 of EB-based differentiation for cyiPS 1–3 and the cyESC line MF12.
First beating areas appeared on day 9 of differentiation. Results of RT-PCR analyses indicated early mesodermal formation with upregulation of Brachyury, followed by induction of cardiac mesoderm with expression of FOG2, NKX2-5, TBX5, and TBX20. Finally, MLC2a and αMHC expression indicated appearance of more mature cardiac cells on day 7 of differentiation (Fig. 3). Even though the cells were assessed in a semiquantitative manner, the results of RT-PCR suggest an upregulation of all tested markers for mesoderm, cardiac mesoderm, and mature cardiomyocytes (CM) during differentiation of cyiPSCs that was similar to cyESCs. Figure 6A shows cross-striations in cyiPS cell–derived myocytes visualized through anti-cardiac troponin T and α-actinin staining. The observed cell membrane associated localization of the gap junction protein connexin 43 between cyiPS cell–derived troponin Tpos CMs (Fig. 6A) suggests functional coupling of these cells.

CyiPSC-derived cardiomyocytes on day 21 of differentiation express typical cardiac proteins and show typical pharmacological responsiveness. (
Electrophysiological characterization of the cyiPS cell–derived CMs was performed using multielectrode arrays. In contrast to smooth muscle and skeletal muscle cells, cardiac myocytes express a functional β-adrenergic pathway and show a positive chronotropic response to β-adrenergic agonists (Schwanke et al., 2006a). Indicative for the presence of an intact β-adrenergic signaling cascade, application of the β-sympathomimetic drug isoproterenol (ISO) at a concentration of 2.5 μM increased the beating frequency of cyiPSC-derived myocytes as shown in Figure 6B.
Also, quinidine, a class IA antiarrhythmic agent and a sodium channel inhibitor, had an effect on FP morphology. As already demonstrated for human iPSC-derived cardiomyocytes (Tanaka et al., 2009), quinidine, also a class IA antiarrhythmic, led to a reduced FP amplitude (Fig. 6B).
Finally verapamil, as a class IV antiarrhythmic drug and an L-type calcium channel blocker, was applied. Comparable to human ESC-derived cardiomyocytes (Liang et al., 2010), verapamil reduced the action potential duration (FDP; time period from the first negative peak (left arrowhead) to the first positive peak (right arrowhead)] and decreased FP frequency (Fig.6B, right) (Liang et al., 2010).
Straightforward generation of stably GFP-expressing cyiPSC clones for development of cellular therapies and preclinical non-human primate models
In our study, we have combined and optimized protocols for single-cell dissociation of hPSCs (Ikeda et al., 2007; Olmer et al., 2010) with prominent advantages provided by the Neon™ Transfection System. The result was a rapid and efficient process, which is directly applicable to conventional feeder-based cultures and does not require time-consuming preadaptation to feeder-free cultures. Applying this optimized protocol, cells of cyiPS clone 2 grown in feeder-based cultures were dissociated into single cells and transfected with the plasmid pCAGGS2 containing GFP under control of the CAG promoter. Nine days after electroporation, the cells were fluorescence-activated cell sorted for GFP-expressing cells. After reseeding, 16 GFPpos colonies were manually picked and cultivated clonally on irradiated MEFs. The 16 colonies showed different growth velocity and different intensities of GFP expression. Of these 16 colonies, six could be expanded and used for further analysis. We observed different intensities and homogeneities in GFP expression between the undifferentiated clones. Only two clones showed homogeneous and strong transgene expression during differentiation (Fig. 7, upper pictures, clone 7). In the other clones, considerable silencing of GFP became visible during differentiation and mosaic-like GFP expression was observed (Fig. 7, lower pictures, clone 3).

Generation of transgenic cyiPSC clones with stable GFP expression in undifferentiated cells and differentiated progeny. As examples, two GFP-transgenic clones with different characteristics are shown. In the upper three rows, images of the cyiPS clone 7 in an undifferentiated and differentiated state are depicted. Notably, clone 7 shows uniform GFP expression after 9 passages of cultivation in undifferentiated cells and after 16 days of differentiation without any signs of silencing. In the lower three rows, images of cyiPS clone 3 are shown. Clone 3 showed a mosaic expression of the transgene GFP in an undifferentiated and differentiated state. Scale bars represent 100 μm. Color images available online at www.liebertpub.com/cell
Discussion
The vast majority of human iPSC lines have been generated using γ-retroviral or lentiviral vectors. The use of common γ-retroviral vectors for reprogramming has various advantages. However, these vectors typically rely on their endogenous retroviral promoter, which might result in inadequate expression levels of the distinct reprogramming factors or might be suboptimal for specific primary cell types to be reprogrammed. In contrast, classical lentiviral vector systems offer the possibility to choose exogenous promoters that confer optimal expression of different reprogramming factors in specific target cell types. Accordingly, HIV-1-based lentiviral vectors encoding different reprogramming factors under control of various promoters have been used successfully for induction of pluripotency in miscellaneous human primary cell types.
HIV-1-derived vectors, however, are not suitable for transduction of many nonhuman primate cells due to an intracellular block for HIV-1 infection with TRIM5α and APOBEC3G as presumed main intrinsic cellular factors (Besnier et al., 2002; Cowan et al., 2002; Munk et al., 2002; Nakayama et al., 2006; Owens et al., 2003; Rahm et al., 2011). Accordingly, all cynomolgus iPSC lines published so far have been reprogrammed using γ-retroviral vectors. Aiming at the lentiviral induction of cynomolgus monkey pluripotent stem cells, we have now used a SIVmac-based lentiviral vector system published by Mangeot et al. (2002). This vector was combined with a previously developed shuttle system (Gruh et al., 2005) to facilitate straightforward construction of expression vectors. In accordance with previous reports (Hanazono et al., 2003), a conventional HIV-1-derived vector proved unsuitable for transduction of cynomolgus monkey fibroblasts, whereas highly efficient transduction of cynomolgus cells was achieved with a SIVmac-based reporter vector, resulting in almost 60% GFPpos cells for a MOI of 1.
A novel “all-in-one” SIV-based reprogramming vector was constructed and successfully applied to simplify reprogramming of M. fascicularis skin fibroblasts. The calculated efficiency of iPSC generation in our experiments was 0.003%. This is comparable to or higher than values previously reported by other groups using retroviral vectors. Deleidi at al. reported an efficiency of 0.005%, Okahara-Narita et al. could obtain several colonies but did not report the calculated efficiency, and Okamoto and Takahashi achieved an efficiency of 0.00005%, only (Deleidi et al., 2011; Okahara-Narita et al., 2012; Okamoto and Takahashi, 2011).
Notably, the established cyiPSCs show a high similarity compared to cyESCs with regard to the typical flat colony morphology and the morphology of individual cells. Standard hESCs/hiPSCs culture conditions were directly applicable, including medium composition and the splitting protocol. In general, cyiPSCs expressed high levels of endogenous ESC marker genes, including OCT4, SOX2, and NANOG similar to hiPSCs and cyESCs as detected by quantitative real-time PCR. This was confirmed through immunocytology, demonstrating expression of common pluripotency markers such as OCT4, SOX2, NANOG, SSEA-4, Tra-1-60, and Tra-1-81 in all cyiPSC clones. Interestingly, the cyiPSCs were negative for SSEA3, which is in good correlation to published data on cyESCs (Suemori et al., 2001).
In addition to detection of dTomato fluorescence by microscopy, transgene expression was assessed applying highly sensitive real-time RT-PCR. Because it has recently been demonstrated for 2A sites that the amount of linked proteins is nearly equal (Szymczak and Vignali, 2005), gene expression and potential silencing of all four reprogramming factors can be determined through analysis of one factor, only. Accordingly, we determined transgene silencing in cyiPSCs via the first transgene OCT4. In good correlation with the observed lack of visible dTomato fluorescence, and in correspondence to recent data concerning the silencing of the SFFV promoter used (Warlich et al., 2011), OCT4 transgene expression appeared to be considerably lower than expression of endogenous OCT4 in all clones. In accordance with previous reports on lentiviral reprogrammed hiPSCs (Haase et al., 2009; Papapetrou et al., 2009; Zhang et al., 2011), however, we detected some degree of variation in the remaining low-level transgene expression in different cyiPSC clones. Despite slightly varying low levels of remaining transgene expression in cyiPSC clones 1 and 3, these clones could be differentiated in vitro and in vivo into derivatives of all three germ layers, albeit at somewhat delayed differentiation rates with slightly lower efficiency as compared to the cyiPS clone 2.
Finally, cardiac differentiation was assessed in more detail for all three clones analyzed. We could detect all stages of cardiomyocyte maturation during differentiation of the cyiPSCs. The three cyiPS cell clones exhibited a different affinity to differentiate into cardiomyocytes. Under the applied differentiation conditions, the number of EBs containing beating areas varied from 9% (cyiPS1) to 17% (cyiPS2). In accordance with the observed synchronous beating of cardiomyocyte clusters, the cardiomycytes stained positively for connexin 43, indicating electrical coupling via gap junctions. Furthermore functional β-adrenergic signaling as a specific feature of cardiomyocytes was proven through stimulation via isoproterenol resulting in a significantly increased beating frequency. The antiarrhythmic drugs verapamil and quinidine showed the expected change in FP duration and beating frequency. This clearly indicates the expression of functional ion channels, an important characteristic of functional cardiomyocytes.
In conclusion, we have constructed an optimized “all in one” SIVmac-based reprogramming vector for efficient transduction and reprogramming of Old World monkey cells. This vector has been successfully used for generation of iPSCs from cynomolgus monkey fibroblasts. These cells show a high degree of similarity to human pluripotent stem cells and differentiate into derivatives of all three germ layers, including functional cardiomyocytes. Cynomolgus iPSCs, and especially the generated stable reporter lines, will allow the establishment of iPSC-based therapeutic concepts in preclinical nonhuman primate models of human physiology and disease. Moreover, these cells will enable the investigation of pluripotency of primate iPSCs in terms of chimera formation and will represent an important tool for generation of transgenic animal models.
Footnotes
Acknowledgments
The authors are grateful to R. Chen, M. Tomala, and T. Scheper for providing recombinant bFGF, A. Kirschning and G. Dräger for providing Y-27632, François-Loïc Cossett and Didier Nègre, École Normale Supérieure de Lyon, France for providing the SIVmac-vector pSIVRMESGAE, and Covance for providing us skin biopsies from cynomolgous monkeys. This work was supported by the German Federal Ministry of Education and Research (01GN0816) as well as by the Excellence Cluster ‘REBIRTH’.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.