
Abstract
Abstract
Porcine induced pluripotent stem cells (iPSCs) are an important animal model for development of regenerative therapies in human medicine. To date, the majority of the porcine cell lines with iPSC characteristics have been generated with the use of viral vectors harboring human or mouse reprogramming factors. Here, we report on the use of Sleeping Beauty transposon vectors based on the porcine transcription factor sequences to reprogram porcine fetal fibroblasts into iPSC-like cells. By using different promoters to drive transgenic expression, we show that the efficiency of reprogramming varies with the promoter type. The cells transfected with two different vector systems under the control of doxycycline-induced tet operator (TetO) promoters failed to upregulate essential endogenous pluripotency genes and to maintain stable iPSC morphology, whereas with the Ef1a and CAG promoters the same vectors proved efficient in generating iPSC-like cells with high levels of endogenous pluripotency gene expression that could be maintained long term in vitro. Our results suggest that the choice of expression vector promoters could significantly influence the efficiency of iPSC production from porcine somatic cells.
Introduction
Due to some known, as well as possibly many unknown, risks associated with the transplantation of pluripotent cells or their derivatives into the human body, iPSCs need to be tested in appropriate large animal models. The domestic pig is a particularly valuable animal model because general physiology and metabolism are similar in human and pigs. The derivation of reprogrammed porcine cell lines with iPSC characteristics has been reported by a number of groups starting in 2009 (Esteban et al., 2009; Ezashi et al., 2009). Most of these lines have been generated with the use of retroviral or lentiviral vectors with integrated human (Ezashi et al., 2009; Telugu et al., 2010; West et al., 2010; Wu et al., 2009) or mouse (Esteban et al, 2009; Montserrat et al., 2011) pluripotency transcription factors. Another method for integration of foreign DNA into the host's genome is by using transposon vectors (Ivics and Izsvak, 2006), which combine the high efficiency of integration, better safety for use compared with viruses, and the possibility to remove the transgenes from the host genome. Here, we report on the reprogramming of pig fetal fibroblasts (pFFs) with Sleeping Beauty (SB) transposon vectors that contain the porcine cDNA sequences coding for OCT4, SOX2, C-MYC, KLF4, and NANOG, in addition to the human LIN28. By using different promoters to drive the transgene expression, we show that the choice of promoter sequence significantly affects the outcome of the reprogramming experiments with porcine somatic cells.
Materials and Methods
Unless specified otherwise, the reagents used were purchased from Sigma-Aldrich.
Construction of SB transposon vectors
The porcine cDNA sequences for OCT4 and NANOG were amplified by reverse transcription (RT)-PCR using mRNA from porcine genital ridge cells from embryos at day 27 of gestation, whereas SOX2, C-MYC, and KLF4 were cloned from pFFs. The human LIN28 sequence was subcloned from plasmid Ef1a_LIN28_ires_Puro (Addgene plasmid 18921; Mali et al., 2008). The cDNA fragments were subcloned into the SB plasmid pT2/HB (generously provided by Dr. Zoltan Ivics, Paul Ehrlich Institute, Langen, Germany) to generate various transposons. Two SB-TetO-pOSMK-ires-dTomato and SB-CAG-pOSMK-ires-Puror vectors (Supplementary Fig. 1) (Supplementary Data are available at www.liebertpub/cell/) were constructed by standard cloning techniques by connecting the transcription factor sequences with P2A, T2a, and E2A self-cleaving viral peptides. Additionally, we constructed SB-TetO-rtTA-pOCT4-ires-dTomato, SB-Ef1a-pOCT4-ires-Puror, SB-TetO/Ef1a-pSOX2-ires-Neor, SB-TetO/Ef1a-pC-MYC-ires-Puror, SB-TetO/Ef1a-pKLF4-ires-Puror, SB-TetO/Ef1a-pNANOG-ires-hLIN28, and SB-CMV-rtTA-ires-Neor (Supplementary Fig. 1). For integration of the transposon into the cellular genomes, we used plasmid pCMV (CAT) T7-SB100X (provided by Dr. Zoltan Ivics), which contains the SB transposase, for co-transfection with the transposon plasmids.
Cell culture
pFFs harboring the mouse Oct4-GFP reporter transgene were isolated from conceptuses at day 27 of gestation (Nowak-Imialek et al., 2011) and were cultured in fibroblast medium [Dulbecco's modified Eagle medium (DMEM) supplemented with 0.1 mM β-mercaptoethanol, 1× nonessential amino acids, 1× penicillin-streptomycin, 2 mM
The transfected cells were cultured initially on gelatin-treated dishes with fibroblast medium, passed on six-well plates with MEF feeders on day 2 posttransfection, and maintained in porcine iPSC medium (DMEM with 0.1 mM β-mercaptoethanol, 1× nonessential amino acids, 1× penicillin-streptomycin, 2 mM
RT-PCR and real-time PCR
RNA was isolated using TRI reagent (Invitrogen), and reverse transcription was carried with MuLV Reverse Transcriptase (Applied Biosystems). PCR was performed with Platinum Taq polymerase (Invitrogen), according to the manufacturer's protocol. The primers used for the amplification of the endo- and transgenes are shown in Supplementary Table 1. To be able to amplify specifically the endogenous transcription factors, the PCR primers were designed to bind to the 3′- untranslated region (UTR) (OCT4, SOX2, C-MYC, KLF4), or to the 5′-UTR (NANOG), as these sequences were not included in the transposon vectors. An IRES reverse primer together with a transgene-binding forward primer was used for determining the presence of the transgenes.
Real-time PCR was carried with Power SYBR Green Master Mix (Applied Biosystems) on ABI 7500 Fast System, according to the manufacturer's protocols. The data were analyzed using GeneX software (bioMCC). Significant differences (where p<0.05) were determined by t-test.
DNA methylation analysis
The methylation status of OCT4 and NANOG promoters was determined by bisulfite sequencing using the EZ DNA Methylation-Direct Kit (Zymo Research) according to the manufacturer's instructions. The identification of CpG islands in the OCT4 and NANOG promoters and the primer designs were performed with MethPrimer (Li and Dahiya, 2002). The primer sequences are shown in Supplementary Table 1.
In vitro differentiation
The iPSC colonies were detached from the feeders by incubation with phosphate-buffered saline (PBS) followed by gentle pipetting, trypsinized, and disaggregated to single cells. Up to 5000 cells were cultured in 25-μL hanging drops in fibroblast medium for 1 week to induce EB formation. The EB-like aggregates were then transferred in gelatinized dishes and cultured for additional 3 weeks. The cells were collected for gene expression analysis. The expression of TE markers CDX2, PAG, and HAND1 was analyzed using primers described by Ezashi et al. (2011).
Results and Discussion
Experiment 1
Transfection of pFFs with SB transposons containing TetO promoters (TetO group) did not result in any iPSC-like colony formation. When MEFs were transfected with SB-TetO-pOSMK-ires-dTomato and SB-CMV-rtTA-ires-Neor, we obtained multiple mouse iPSC-like colonies, which downregulated the transgenes in spite of the continued DOX supplementation (as verified by loss of dTomato fluorescence and RT-PCR). These cells also upregulated a number of endogenous pluripotency markers, such as AP, SSEA-1, Oct4, Sox2, Nanog, Zfp42, and Utf1, and ultimately could be differentiated in vitro into cells from the three germ layers, including neuronal-like cells, epithelial-like cells, and rhythmically contracting cardiomyocytes (results not shown). These results suggested that the porcine fibroblasts may have different requirements for reprogramming to pluripotency compared with mouse cells, such as different culture conditions, need for other reprogramming factors, or a requirement for higher expression levels of the transgenes. Although culture conditions used for mouse or human ESCs have been insufficient for maintaining pluripotency in ungulate ESCs (cell morphologies and culture conditions have been reviewed in detail by Nowak-Imialek et al., 2011), they have been adequate for the derivation of porcine iPSC cells (Esteban et al., 2009; Ezashi et al., 2009). The sustained expression of the exogenous pluripotency transcription factors that has been reported in these cells could not be excluded as an important factor in the maintenance of their pluripotency characteristics.
The types of transgenes we used have also been proven to be sufficient in the derivation of porcine iPSC (Esteban et al., 2009; Ezashi et al., 2009). Therefore, the transgene expression level was considered to be the more likely cause for the absence of iPSC-like colonies. Because the TetO promoter has been reported to drive lower expression in primary fibroblasts compared with the constitutive promoters (Qin et al., 2010), we reasoned that this could be corrected by using the more powerful Ef1a or CAG promoters. To check this hypothesis, we transfected the same pFF lines with transposons from the CAG-Ef1a group. Within 2 weeks posttransfection, we obtained multiple compact colonies [mean=2195; standard error of the mean (SEM)=166; n=3] that were 95% AP-positive, and approximately 70–80% had upregulated the OCT4-GFP reporter (Fig. 1A, B). Immunocytochemistry revealed the absence of cells positive for SSEA-1 (Fig. 1C–D), while SSEA-4 stained only some colonies (Fig. 1E–F) as well as many single cells with fibroblast-like morphology. SSEA-4 expression did not correlate with the OCT4-GFP reporter expression. We picked green fluorescent protein (GFP)-positive colonies and expanded them further to derive iPSC-like cell lines (Fig. 1G) that proliferated and maintained mouse iPSC-like morphology and OCT4-GFP expression (Fig. 1H) for over 37 passages. These cells showed expression of endogenous OCT4, SOX2, NANOG, REX1, CDH1, and TDH (Fig. 2A). However, transgene expression from the transposons was not silenced (Fig. 2B).
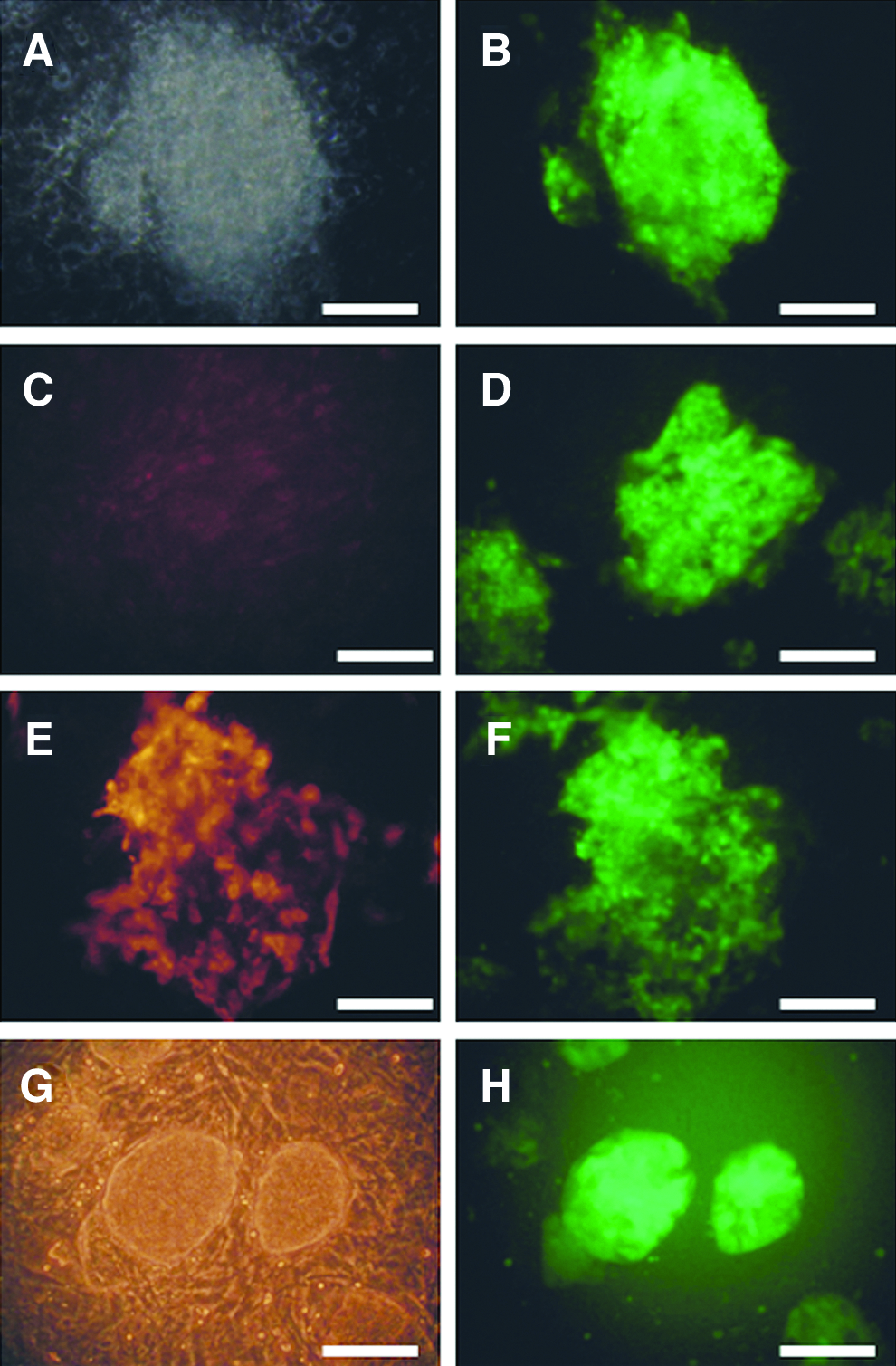
Porcine iPSC-like cells produced in Experiment 1. (
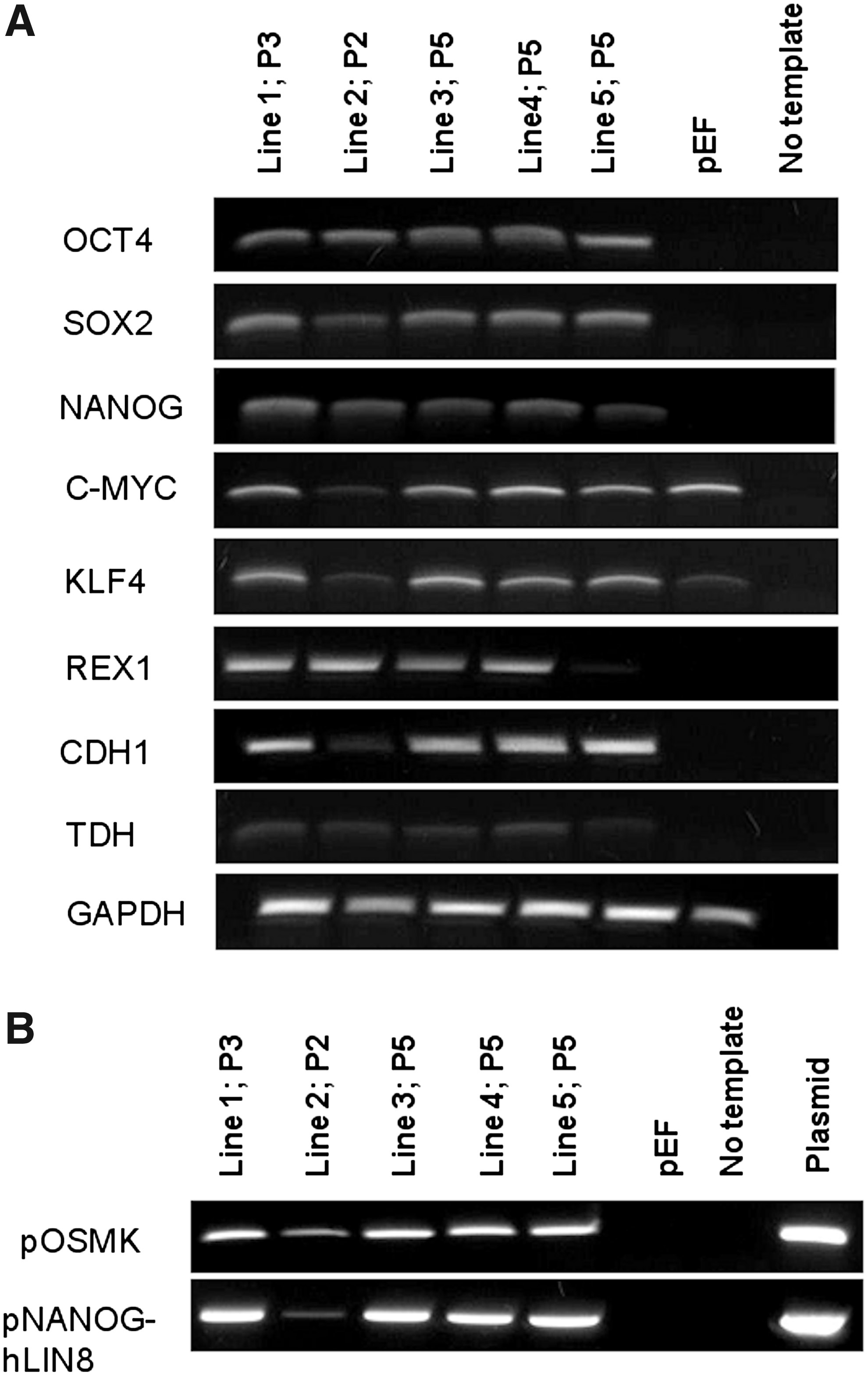
Expression of endogenous pluripotency genes (
Experiment 2
The transfection of pFFs with the two different promoter groups (TetO group and Ef1a group) resulted in multiple compact iPSC-like colonies in both groups; however, although there was no significant difference in the primary colony numbers, the colonies in the TetO group did not show any OCT4-GFP reporter expression (Fig. 3A–B), had only weak AP activity (Fig.3C) and no SSEA-1 or SSEA-4 activity. On the other hand, in the Ef1a group approximately 15% OCT4-GFP reporter-positive colonies were observed (Fig. 3D–E) that showed uniform strong AP activity (Fig. 3F) and were approximately 8% SSEA-1 positive; however, the SSEA-1 expression did not correlate with the OCT4-GFP reporter expression. Picked colonies from the TetO group formed cell lines that proliferated very fast and required splitting at ratios of 1:20–1:35, but lost iPSC morphology within 7–8 passages. Withdrawal of DOX resulted in cessation of proliferation and/or reversal to fibroblast morphology, suggesting that the transgenes were primarily responsible for driving cell growth.
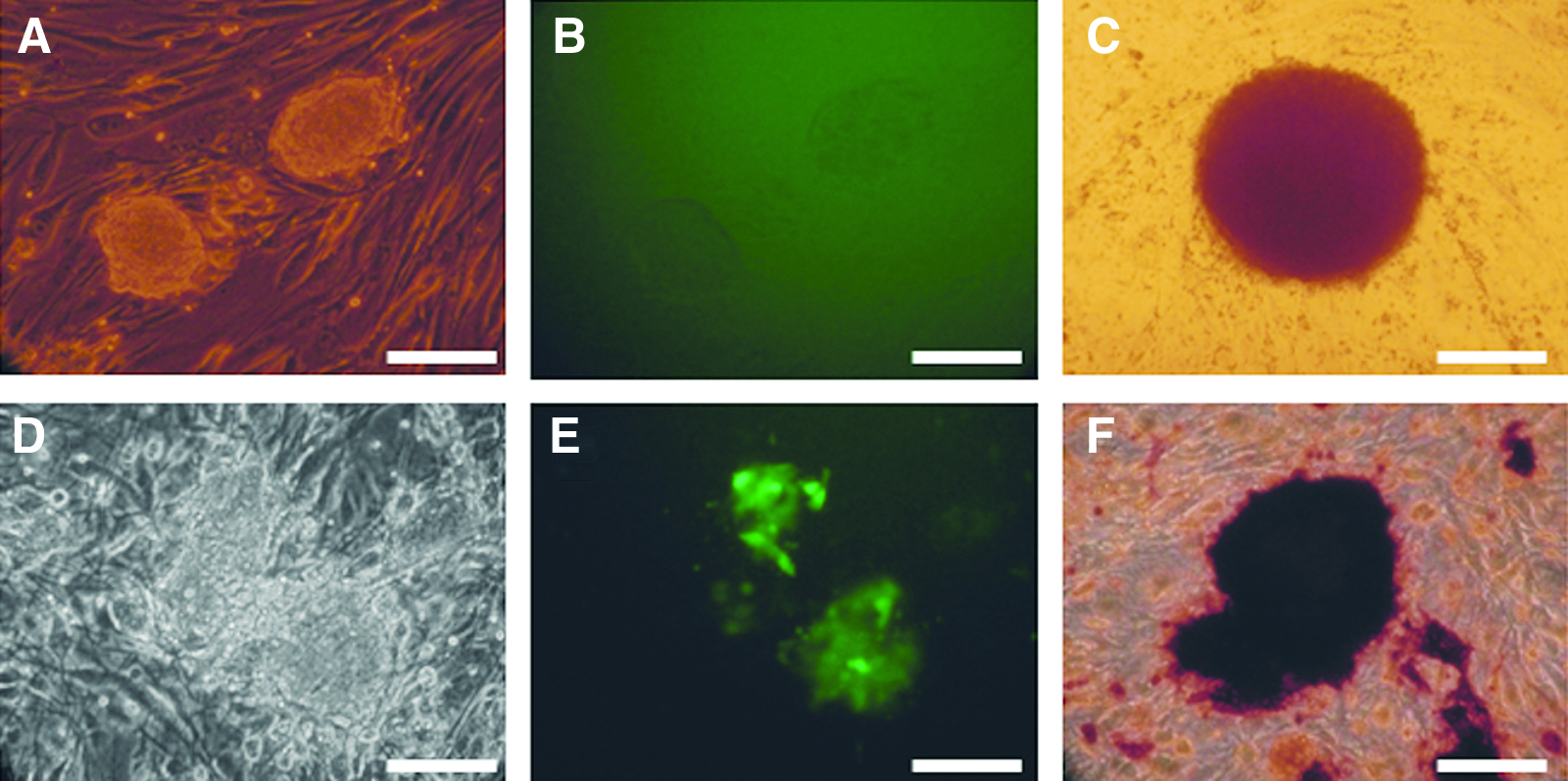
Porcine iPSC-like cells produced in Experiment 2. In the TetO group, compact colonies (
These results suggest that these cells were only induced to proliferate, but were not reprogrammed to pluripotency. On the other hand, picked GFP-positive colonies from the Ef1a group proliferated for at least 20 passages and maintained iPSC-like morphology; however, many cells lost reporter fluorescence after P10. Overall, the efficiency of generating OCT4-GFP reporter-positive iPSC-like colonies in this group was lower compared with the CAG-Ef1a group in Experiment 1, likely due to an effect of transgene arrangement in the expression vectors.
Our experiments showed that each promoter type may play a different role in the reprogramming, possibly by modulating the expression levels of the transgenes, which could affect the activation of the endogenous pluripotency genes. To investigate whether there are differences in the endogenous as well as transgene expression levels between the different promoter groups, we performed relative quantitative real-time PCR analysis in three primary cultures from the CAG-EF1a group in Experiment 1 and in each promoter group from Experiment 2. The results from the endogenous gene expression analysis are summarized in Figure 4. The relative expression levels of OCT4, SOX2, NANOG, REX1, and CDH1 were significantly higher in the CAG-Ef1a group compared to both groups in Experiment 2, whereas there was no difference in the expression of TDH compared with the TetO group. Within Experiment 2, the relative expression of OCT4, NANOG, and CDH1 was significantly higher in the Ef1a group compared with the TetO group, which did not vary significantly from pFF. The relative expression of SOX2 was similar in both groups, whereas TDH expression was higher in the TetO group. The expression of C-MYC and KLF4 was not significantly different in any of the three iPSC-like groups and was lower compared with pFF, which is in agreement with results reported by Ezashi et al. (2009).
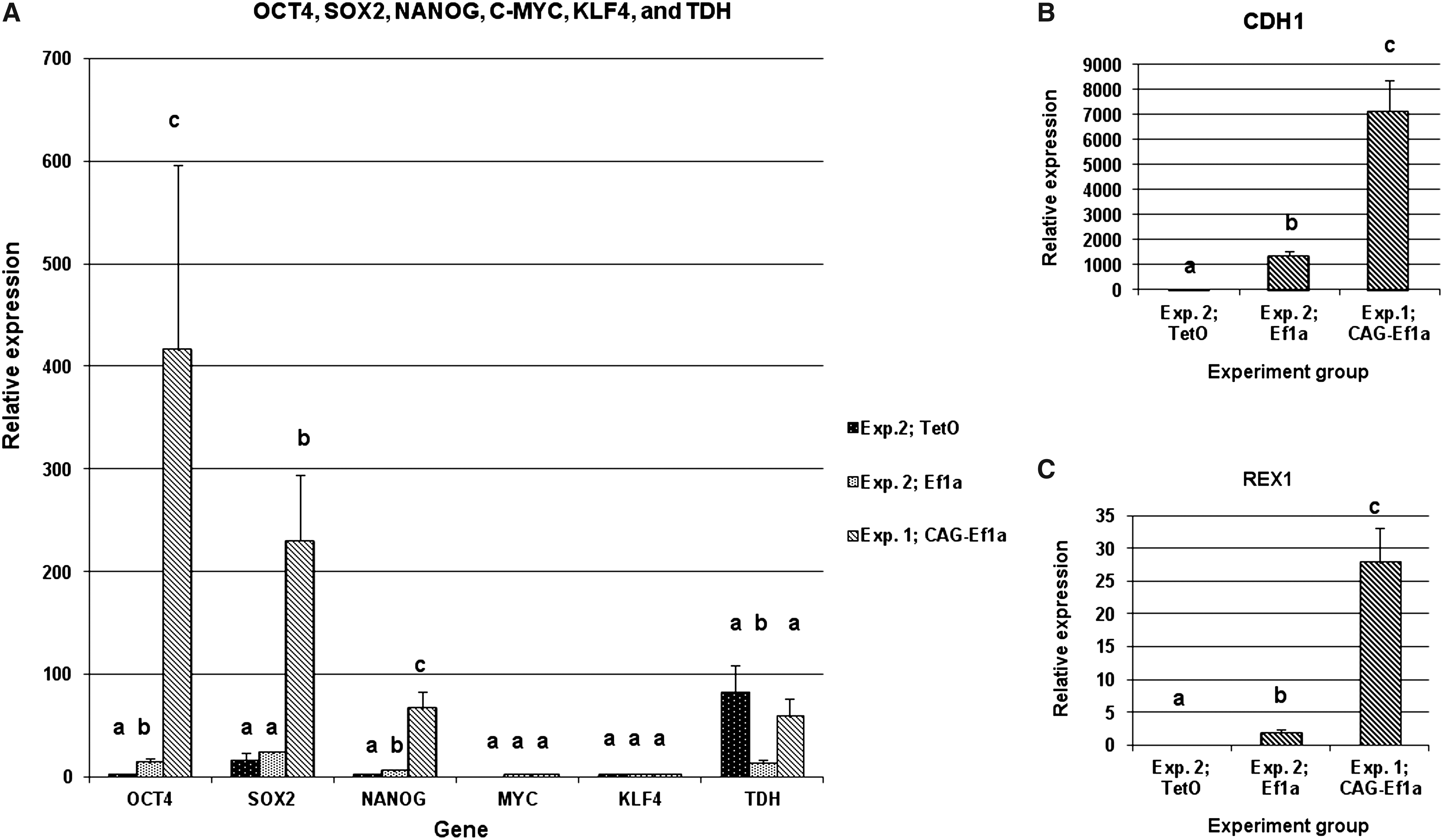
Relative quantitative real-time PCR analysis of endogenous pluripotency gene expression in porcine iPSC cells produced with different promoters. (
In our analysis of transgene expression, we compared mainly the TetO and Ef1a groups in Experiment 2, because in Experiment 1 the TetO group did not provide any colonies for analysis. Only the expression of trans-KLF4 and trans-NANOG-LIN28 in the CAG-Ef1a group was compared with the results from Experiment 2, due to the fact that the primers used and their corresponding amplification sequences were identical in all groups. The results are shown in Figure 5. There was no difference in the expression of trans-OCT4 and SOX2 between the TetO and Ef1a groups, whereas trans-C-MYC expression was significantly higher in the TetO group. On the other hand, the expression of trans-KLF4 and trans-NANOG-LIN28 in the Ef1a group and in the CAG-Ef1a group was significantly higher compared with TetO. This suggests that the high expression of these transgenes might be important for the reprogramming of porcine somatic cells to iPSC. Including NANOG-LIN28 seemed necessary for the production of iPSC-like colonies in Experiment 1, whereas it was dispensable for colony formation in Experiment 2; however, no OCT4-GFP expression was detected when this construct was omitted (results not shown).
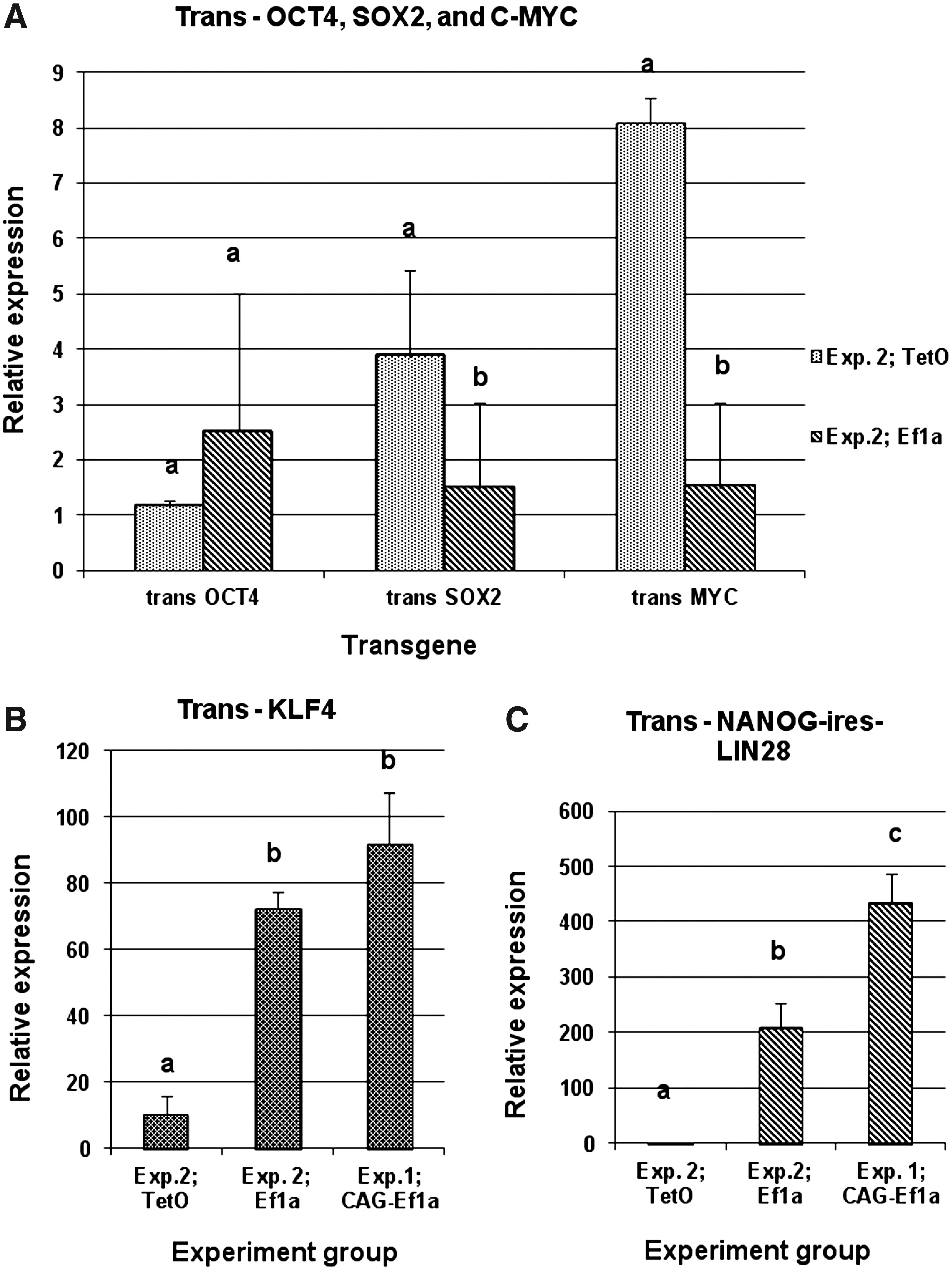
Relative quantitative real-time PCR analysis of transgene expression in porcine iPSC cells produced with different promoters. (
It was surprising that the transgenic OCT4 and SOX2 expression levels were similar in the TetO and Ef1a groups, because the TetO promoter is considered to drive lower levels of transcription compared with constitutive promoters (Qin et al., 2010). The similarity in expression levels of trans-OCT4 could be explained with the design of the vector, where the rtTA expression cassette is ligated in front of the OCT4 promoter and may additionally stimulate downstream transcription. This is supported by the fact that iPSC-like colonies did not form when the rtTA expression cassette was on a separate plasmid (results not shown). The unexpected trans-SOX2 and C-MYC expression levels could be explained with selection, where only the cells with certain expression levels of these genes can form compact colonies. The high trans-C-MYC could also explain the increased rate of proliferation of the iPSC-like cells in TetO group.
The reprogramming to pluripotency is an epigenetic process that involves alterations in the DNA methylation patterns as well as modification of chromatin epigenetic marks (Hochedlinger and Plath, 2009; Papp and Plath, 2011). To examine the DNA methylation status of the OCT4 and NANOG promoters, we performed bisulfite sequencing analysis in pFFs and in one line from each experiment that expressed the OCT4-GFP reporter. Our results showed that in both lines the OCT4 and NANOG promoters were hypomethylated in comparison with pFF (Supplementary Fig. 2). These results confirm that epigenetic reprogramming had taken place in the derived iPSC-like lines.
The ability of the iPSC-like cells to differentiate into different cell types was tested in vitro by EB formation in “hanging drops” followed by culture on gelatin-coated plastic dishes. Although EB-like spheres were readily formed (Supplementary Fig. 3A), the subsequent culture resulted in the formation of limited number of cell types, mainly trophoblast-like and fibroblast-like cells, and in some cases, neuronal-like outgrowths. Many of the cells that proliferated from the EB-like formations retained iPSC morphology and strong OCT4-GFP reporter expression (Supplementary Fig. 3B–C). The trophoblast-like cells, identified by formation of large vesicles (Supplementary Fig. 3D) and by the upregulated expression of CDX2, PAG, and HAND1 (Supplementary Fig. 4), failed to downregulate the endogenous pluripotency genes as well as the transgenes (Supplementary Fig. 4). These results suggest that the limited differentiation abilities of the iPSC-like cells could be due to a lack of full reprogramming to pluripotency, or the continuous expression of the transgenes, or to suboptimal culture conditions. It has been shown that porcine blastocysts express OCT4 in both inner cell mass and trophectoderm (Kirchhof et al., 2000); therefore, it is possible that the persistent transgene expression does not hinder trophoblast differentiation, but limits the differentiation into other lineages. Currently, we are exploring the possibilities of removal of the transposons from the cell genomes in addition to optimizing the culture conditions.
In conclusion, we demonstrate that the choice of expression vector promoter may affect the outcome of the reprogramming experiments with porcine somatic cells, possibly by modulating the expression levels of the reprogramming transcription factors. The optimal conditions for generation of porcine and mouse iPSCs may be different not only in terms of culture conditions, but also in terms of transgenic expression levels. Our data suggest that relatively higher expression of OCT4, KLF4, and NANOG or LIN28 versus SOX2 and C-MYC may improve the induction of pluripotency-related genes in porcine iPSC production.
Footnotes
Acknowledgments
We thank Dr. Zoltan Ivics from Paul Ehrlich Institute, Langen, Germany, for providing the Sleeping Beauty transposon/transposase plasmids and for his technical advice. This study was supported by a grant by the German Research Foundation (Deutsche Forschungsgemeinschaft) Ni 256/ 32-1, by the Danish National Advanced Technology Foundation, and by EU FP7 projects PartnErS (PIAP-GA-2008-218205) and PluriSys (HEALTH-2007-B-223485).
Author Disclosure Statement
The authors declare that there are no existing conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.