
Abstract
Abstract
The production of healthy, live, cloned animals by somatic cell nuclear transfer (SCNT) has been hampered by low efficiencies. Significant epigenetic changes must take place to ensure proper chromatin remodeling in SCNT. We hypothesized that exogenous expression of OCT4 in donor fibroblasts prior to its fusion with enucleated oocytes would facilitate SCNT reprogramming. We infected bovine adult fibroblasts with retroviral vectors containing yellow fluorescent protein (YFP) only, or the OCT4 gene fused to YFP (YO). We found that development to the blastocyst stage was not different between NT-YFP and NT-YO groups. NT-YFP embryos had the fewest trophoblast cells, measured by numbers of CDX2-positive cells. Fibroblasts expressing OCT4 had reduced levels of histone 3 lysine 9 or 27 trimethylation (H3K9me3 and H3K27me3, respectively). NT-YO blastocysts displayed higher H3K9me3 levels than IVF and NT-YFP embryos; however, they did not have different H3K27me3 levels. Levels of XIST mRNA expression in NT-YO and NT-YF were higher when compared to in vitro–fertilized blastocysts. We observed no differences in the expression of SOX2, NANOG, and CDX2. Although overexpression of OCT4 in donor cells increased H3K9me3 and did not reduce XIST gene expression, we show that a single transcription factor can affect the number of trophectoderm cells in bovine SCNT embryos.
Introduction
Cellular reprogramming in SCNT involves changes in gene expression that are associated with epigenetic modifications (Latham, 2005). Chromatin modifications, including DNA methylation, histone lysine methylation, and histone acetylation, are components of the epigenetic mechanism that ultimately determine the expression level of most genes (Santos and Dean, 2004). Histone 3 trimethylation of lysine 9 (H3K9me3) and lysine 27 (H3K27me3) are associated with transcriptional repression, and its changes are catalyzed by specific histone methylases or demethylases (Hublitz et al., 2009). With the exception of imprinted genes, both paternal and maternal genomes are demethylated in fertilized embryos, and de novo methylation occurs, leading to an asymmetrical pattern that characterizes the inner cell mass (ICM) and the trophectoderm (Santos and Dean, 2004). This pattern is not well observed in bovine embryos derived from SCNT, as seen by homogeneous staining of the ICM and trophectoderm (TE) with antibodies against DNA methylation or H3K9me3 (Santos et al., 2003). In the mouse, H3K27me3 staining was more intense in the ICM of control IVF blastocysts, whereas asymmetry was not seen in SCNT embryos (Zhang et al., 2009). These results suggested that the lower number of live births from SCNT might stem from errors in the reestablishment of histone methylation patterns.
In an attempt to improve bovine SCNT results, a series of studies have tried to induce epigenetic changes either before or after the somatic cells fuse to the oocyte. Using the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza) in bovine donor cells increased the blastocyst rate when compared to nontreated cells (Enright et al., 2005). Histone deacetylase inhibitor trichostatin A (TSA) led to SCNT embryos having similar levels of in vitro development and histone H4 acetylation at lysine 5 to IVF embryos (Iager et al., 2008). The combined use of 5-aza and TSA increased both blastocyst rates (Ding et al., 2008) and development to term (Wang et al., 2011). The use of oocyte extract to induce changes in the donor cell population in vitro has also reportedly reduced histone acetylation, increasing blastocyst formation in cows (Tang et al., 2009), and increasing live birth rates in sheep (Rathbone et al., 2010).
Oocytes contain significant levels of mRNA for the pluripotency-related gene OCT4 (Kocabas et al. 2006; Kurosaka et al. 2004; Schöler et al., 1990). OCT4, part of the POU transcription factor family, binds an octameric nucleotide motif within promoters or enhancer regions (Pesce and Schöler, 2001). In addition to being expressed in oocytes, OCT4 is also expressed in mouse pluripotent embryonic stem cells (ESCs) and primordial germ cells (Schöler et al., 1990). The use of mouse ESCs as donor cells in nuclear transfer increases the number of live births when compared to somatic donor cells (Rideout et al., 2001). OCT4 is generally considered the most important of the reprogramming factors used to produce induced pluripotent stem cells (iPSCs; Takahashi and Yamanaka, 2006; Yu et al., 2007). Generation of iPSCs from neuronal stem cells has been achieved using OCT4 alone (Kim et al., 2009). In addition, suppression of OCT4 in mouse ESCs reduced the expression levels of several chromatin remodeling genes (Sharov et al., 2008), and OCT4-null embryos displayed more compact chromatin in pluripotent epiblast stem cells than wild-type embryos (Ahmed et al., 2010). On the basis of these facts, we hypothesized that the exogenous expression of OCT4 in the donor fibroblast might facilitate SCNT reprogramming by the oocyte.
Our goal was to evaluate the epigenetic and gene expression changes in SCNT-derived bovine embryos using donor fibroblasts ectopically expressing OCT4. In this study, we used a retroviral vector to introduce the human OCT4 gene into the bovine adult fibroblasts being used for SCNT. We characterized epigenetic changes in these fibroblasts and generated SCNT blastocysts. We assessed development rates and analyzed these embryos for ICM and TE cell allocation, for the degree of methylation at H3K9 and H3K27 residues using specific antibodies for trimethylation, and to quantify gene expression of embryonic transcription factors and epigenetic modifying enzymes.
Our results show that OCT4 expression in donor fibroblasts could reduce the global levels of histone trimethylation and increase the expression of demethylases. The SCNT embryos produced using these cells had more TE cells than the control SCNT-derived embryos. Expressing OCT4 in donor cells increased H3K9me3 global levels but did not induce changes in H3K27me3 levels. We found an increase in gene expression of XIST and a reduction in endogenous OCT4 in both SCNT groups as compared to their IVF counterparts.
Materials and Methods
Unless otherwise stated, we purchased all reagents from Sigma (St. Louis, MO).
Production of transgenic cells
The human OCT4 open reading frame was cloned from from human full-length Oct4 IMAGE clone 40125986/MGC151044 using specific primers (Table 1). The 1080-bp Oct4 open reading frame (ORF) PCR product was digested with MluI (5′) and ClaI (3′) and inserted in-frame onto the carboxyl terminus of yellow fluorescent protein (YFP) into the retroviral vector NITSC-YFP, which was digested with the same enzymes to produce the YFP–hOct4 fusion protein expression vector. The vector was packaged by three-way transfection of HEK293 cells using the packaging plasmids cytomegalovirus–glycoprotein (CMV-GP) and CMV–vesicular stomatitis virus G protein (VSVg). At 48 h after transfection, the viral supernatant was harvested, filtered through a 0.45-μm filter to remove floating human embryonic kidney (HEK) cells, and frozen at −80°C in aliquots until use to infect target cells. The Moloney murine leukemia virus (MMLV) retroviral vector contained neomycin selection cassette and Tet-Off regulation, all flanked by a long terminal repeat (LTR) promoter (Fig. 1A). We produced viral vectors containing YFP only (YFP) for control studies or YFP fused to OCT4 (YO), using HEK cells cultured in fibroblast culture medium consisting of Dulbecco's minimum essential medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids (NEAA), and 1% penicillin/streptomycin. We filtered the supernatant of infected HEK cells and then used it to transduce bovine adult fibroblasts (BAFs) from a female Jersey heifer. We thawed the BAFs at the fifth passage and cultured them at low confluency with fibroblast culture medium. We transduced cells using three 8-h rounds of viral medium and then cultured them on DMEM, changing the medium every 5 days. We selected positive cells by adding 500 μg/mL of G418 to the culture for 10 days. Cells were then passaged and cultured with G418 for 5 more days before freezing. Cells were thawed, expanded for two more passages, and frozen again in working aliquots. We checked for the presence of YFP using an inverted fluorescence microscope (Nikon TE-2000, Tokyo, Japan).
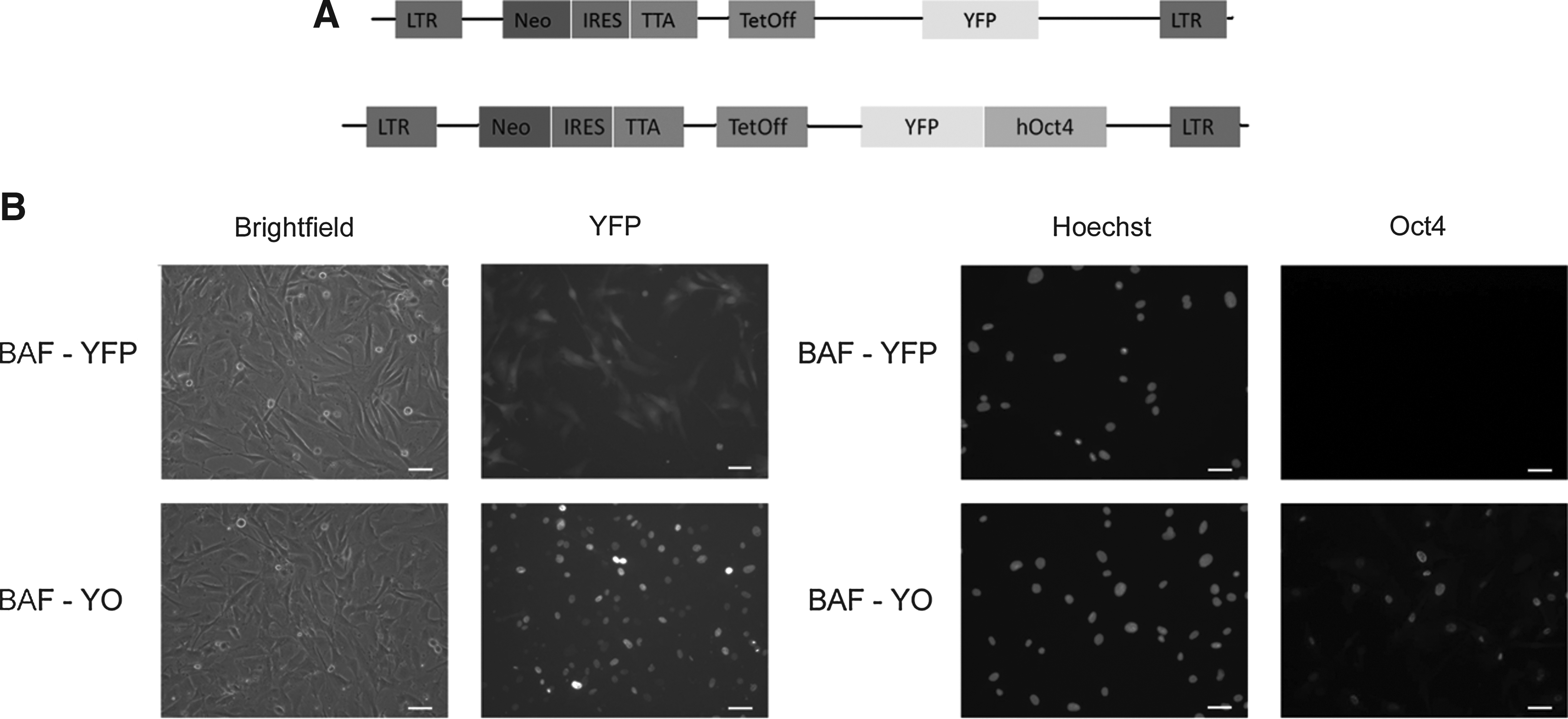
Generation and characterization of transgenic fibroblasts. (
Immunocytochemistry of cultured cells
We used phosphate-buffered saline
In vitro maturation of oocytes
We harvested bovine oocytes from slaughterhouse-derived ovaries, as described previously (Ross and CibelLi, 2010). We placed the oocytes in tissue culture medium-199 (TCM-199) medium containing 10% percent FBS, 3 μg/mL luteinizing hormone (LH; Sioux Biochemical, Sioux Center, IA, USA), 3 μg/mL follicle-stimulating hormone (FSH; Sioux Biochemical), 22 μg/mL sodium pyruvate, and 25 μg/mL gentamicin (Invitrogen) for 16 h for SCNT or 22 h for IVF.
SCNT
We used a bovine SCNT protocol similar to the one previously described (Ross and CibelLi, 2010), with the modifications described below. We cultured YFP or YO donor nucleus fibroblasts in serum starvation (0.5% FBS in DMEM) for 24 h and collected them using a 10-UI/mL Pronase solution. Cells and enucleated oocytes were placed in drops of HECM HEPES medium. Using a glass pipette with 20 μm, we verified YFP-positive cells by brief exposure to fluorescence and individually placed them in the perivitelline space of each oocyte. After reconstruction, we fused, activated, and cultured these oocyte-cell couplets as described in the protocol. After 7 days in culture, blastocysts rates were recorded and used for protein or RNA expression analysis.
In vitro fertilization
We transferred mature oocytes into Tyrode's albumin lactate pyruvate (TALP)-based fertilization medium (Parrish et al., 1986) supplemented with 20 μg/mL heparin. Frozen–thawed semen was fractionated in a Percoll gradient and 1×106 sperm cells/mL were added to the medium containing the oocytes. Fertilization was carried out for 18 h at 38.5°C and 5% CO2 in high humidity. Presumptive zygotes were then denuded and cleaned from excessive sperm using HH medium and cultured in vitro as described above for 7 days.
Immunocytochemistry of blastocysts
All solutions were prepared with PBS. We washed embryos three times in PBS and fixed them with 4% paraformaldehyde solution for 20 min. Then, we washed day-7 (D7) blastocysts three times in 0.1% Triton X-100 solution and permeabilized them for 15 min with 0.5% Triton X-100 solution. We performed the blocking of unspecific binding sites using 0.1% Triton X-100, 1% BSA, and 10% normal donkey serum solution for 1 h at room temperature. We incubated primary antibody for 2 h in the dark at room temperature in 0.1% Triton X-100 and 1% BSA solution. We washed embryos three times for 15 min in 0.1% Triton X-100 solution, followed by incubation with secondary antibody for 1 h at room temperature in the dark. We then washed the embryos three times for 15 min in the dark and mounted them on slides with Prolong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). We visualized the blastocysts using an inverted spinning-disk confocal microscope, and obtained stacks of pictures using Metamorph software (Molecular Devices, Sunnyvale, CA, USA). We used the same primary antibodies as above and anti-CDX2 (sc166830; Santa Cruz Biotechnology). We used the same secondary antibodies as above and Alexa Fluor 568 donkey anti-mouse immunoglobulin G (Invitrogen).
Intensity quantification and differential cell counting
Using Metamorph software for image analysis, we performed intensity quantification of fibroblasts by delineating each nucleus; the software calculated average pixel intensity. Data were normalized by an average of two background regions for each projection. We randomly selected and quantified three different image fields for each group (YFP and YO). For H3K9me3 quantification, 63 YFP cells and 100 YO cells were quantified. For H3K27me3 quantification, 57 YFP cells and 111 YO cells were quantified.
We performed differential cell counting by counting CDX2-positive cells and DAPI-stained nuclei on all planes of images obtained from a single embryo. We considered CDX2-positive cells as TE (Kuijk et al. 2008) and CDX2-negative cells as ICM. We determined the total cell number by DAPI staining. A total of 63 embryos were used: 18 for IVF, 21 for NT-YFP, and 24 for NT-YO.
We performed the intensity quantification for embryos as described previously (Ross et al., 2008). Briefly, all planes were combined in a maximum projection, and each nucleus was assessed individually for its average pixel intensity. Data were normalized, as mentioned earlier for fibroblasts. Each staining used a total of 10 embryos, developed from two different batches of oocytes, for each group (IVF, YFP, and YO).
RNA extraction, reverse transcription, and quantitative PCR
We pooled 5–10 blastocysts per group from each oocyte batch in a total of five replicates. Embryos were placed in 20 μL of extraction buffer from the PicoPure RNA Extraction Kit (Applied Biosystems, Foster City, CA, USA), incubated for 30 min at 42°C, and stored at −80°C until further processing. We thawed samples on ice and extracted RNA according to the PicoPure manufacturer's protocol, including a step for incubation with DNase (Qiagen, Hilden, Germany). Isolated RNA was used for reverse transcription reaction using the Superscript II Reverse Transcriptase Kit (Invitrogen) with 250 ng of random primers (Promega, Madison, WI) following the manufacturer's instructions. We cleaned the cDNA that was synthesized using Zymo Research DNA Clean and Concentrator kit (Zymo Research, Irvine, CA, USA). We quantified the cDNA using a NanoDrop Spectrophotometer (Thermo Scientific, Rochester, NY, USA), and we diluted all samples to 2 ng/mL. We performed duplicate quantitative PCR reactions for each sample, using SYBR Green 2X PCR Master Mix (Applied Biosystems) and 2 μL of cDNA with the ABI 7000 Detection System. Thermal cycle settings were 40 cycles of 95°C for 15 s and 60°C for 60 s. We used the histone 2A (H2A) housekeeping gene to normalize the expression of target genes, using the ΔΔCt method (Pfaffl, 2001). Table 1 describes the primers used.
Statistical analysis
We used SAS software (SAS Institute, Cary, NC) to analyze our data. We performed analysis of variance (ANOVA) using PROC GLM, considering treatment as an independent variable and using Tukey's adjustment as a post hoc test to compare means. We analyzed quantitative PCR data using PROC MIXED from SAS, considering the oocyte batch as a random variable. We set the statistical significance at p<0.05.
Results
Characterization of transgenic cells
In the present study, we generated transgenic BAFs expressing OCT4 fused to YFP (BAF-YOs) or YFP only (BAF-YFPs) for use as donor cells for SCNT. After viral transduction by means of retroviral vectors (Fig. 1A), selection with neomycin, and freezing, we thawed the BAFs and detected YFP fluorescence in the cytoplasm of the BAF-YFP cells and in the nuclei of the BAF-YO cells (Fig. 1B). Immunocytochemistry revealed that OCT4 protein was not expressed by BAF-YFP cells, but, as expected, it was present and properly localized in BAF-YO cells (Fig. 1C).
To determine if OCT4 ectopic expression affected epigenetic markers in fibroblasts prior to nuclear transfer, we performed immunocytochemistry for H3K9me3 and H3K27me3 (Fig. 2A). Analyzing both markers for the intensity of immunofluorescence showed that BAF-YO had significantly less intensity than BAF-YFP cells (Fig. 2B). To further investigate the possible causes, we assessed gene expression of histone modification enzymes known to be regulated by OCT4 in mouse ESCs (Loh et al. 2006). Quantitative RT-PCR showed that the amount of RNA for the enzymes responsible for H3K9 and H3K27 methylation (EHMT1) and demethylation (JARID2) were not statistically different between the two cell lines; however, H3K9 demethylase KDM4C was upregulated in BAF-YO cells (Fig. 2C), suggesting that active demethylation likely accounts for the diminished signal.
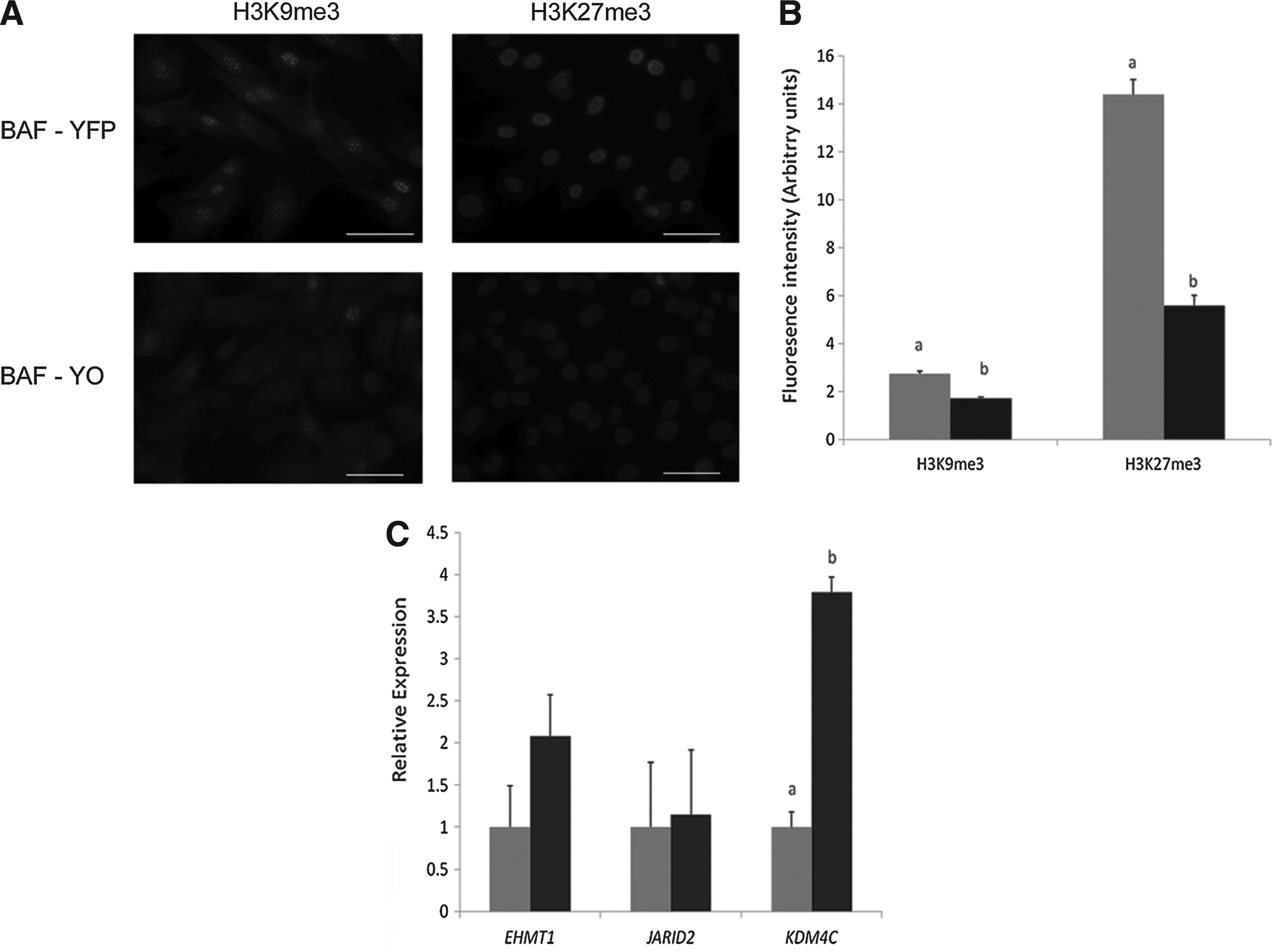
Characterization of transgenic fibroblasts. (
Embryo development rates and differential cell counting
We performed a total of 12 SCNT manipulations in which we assessed fusion, cleavage, and blastocyst rates; we observed no differences between BAF-YFP– or BAF-YO–derived embryos (Fig. 3). Although YFP was clearly visible in the donor cells, we could observe no YFP fluorescence 72 h after SCNT activation in either BAF-YFP– or BAF-YO–derived embryos (henceforth referred to as NT-YFP and NT-YO, respectively). To verify cell allocation to ICM or TE, we performed immunocytochemistry against the CDX2 protein in a total of 63 IVF- and SCNT-derived day-7 blastocysts. Confocal images showed that CDX2 staining was limited to the TE of all IVF or SCNT embryos (Fig. 4). Both SCNT groups had a lower number of ICM cells than the IVF control, although the number of TE cells was higher in both IVF and NT-YO than in NT-YFP embryos (Table 2). Consequently, the total number of cells was higher in IVF blastocysts than in NT-YFP embryos, whereas NT-YO embryos did not statistically differ from either group (Table 2).
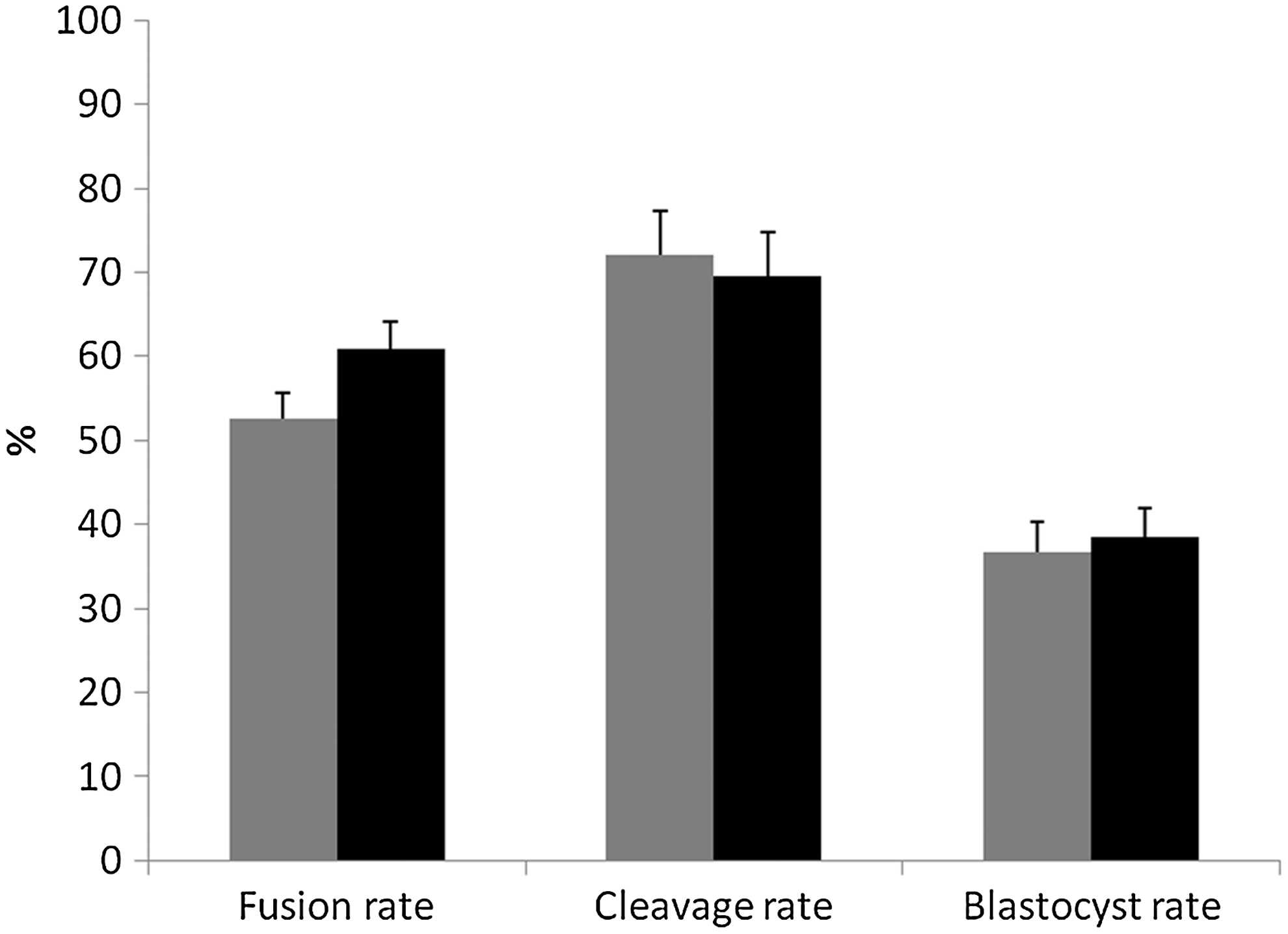
Fusion, cleavage, and blastocyst rates (as a proportion of the total fused structures) of somatic cell nuclear transfer using BAF-YFP (NT-YFP, grey bars) or BAF-YO (NT-YO, black bars) as donor cells. Different superscript letters indicate significant statistical difference (p≤0.05); n=12.
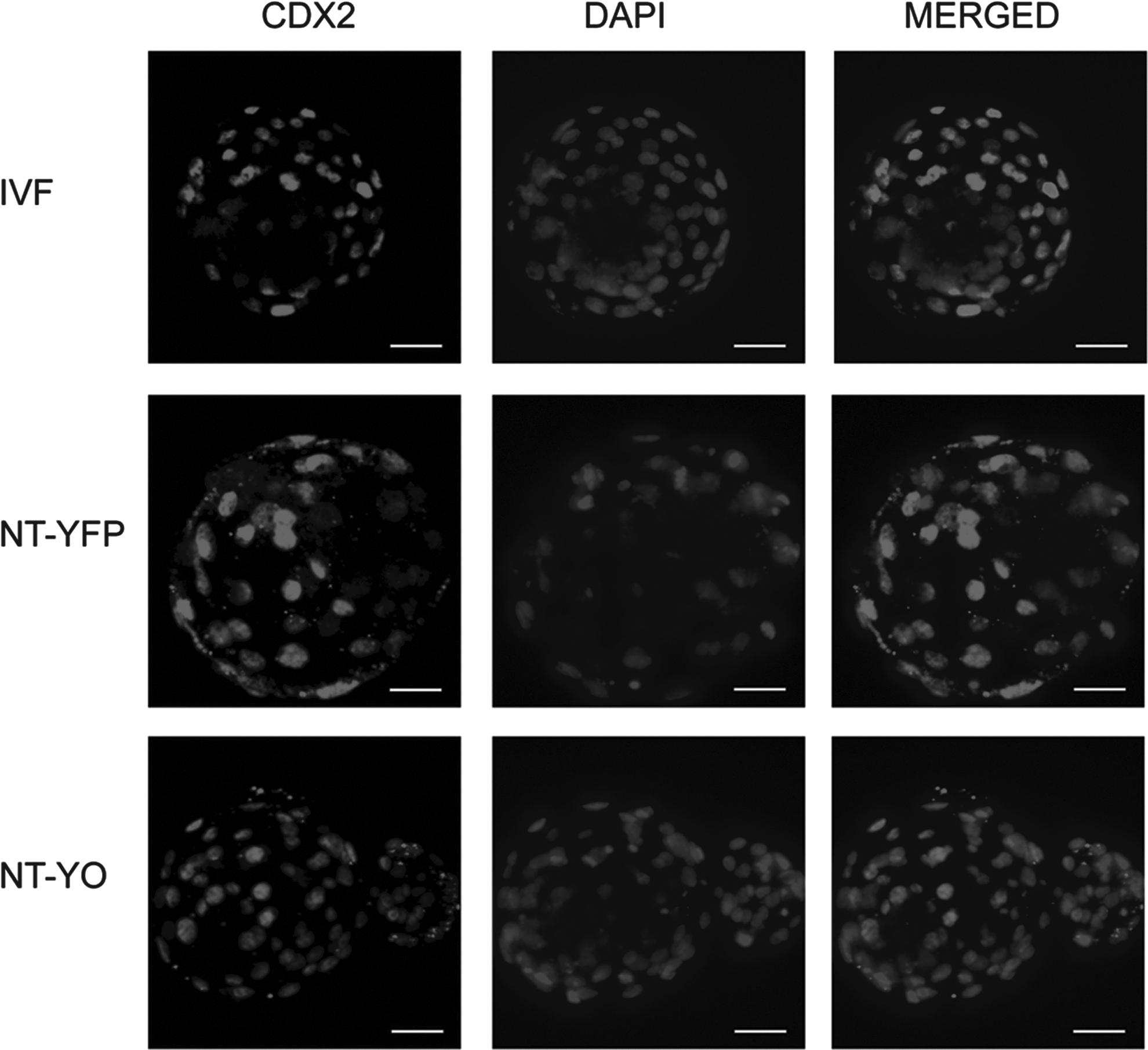
CDX2 immunostaining in IVF, NT-YFP and NT-YO blastocysts. CDX2 protein is present in the nucleus of trophoblast and absent in ICM cells. Scale bars, 40 μm.
Different superscript letters indicate significant statistical differences (p ≤0.05).
We considered positive cells as trophoblast cells and negative cells as ICM cells.
TE, trophectoderm; ICM, inner cell mass.
Histone trimethylation quantification
We performed immunocytochemistry against H3K9me3 and H3K27me3 in IVF, NT-YFP, and NT-YO day-7 blastocysts to study modifications in the chromatin that could be induced by expressing OCT4 in donor cells. We quantified the average intensity using Metamorph software. We observed no obvious difference of intensity in the ICM and TE of H3K9me3-stained embryos (Fig. 5A); however, NT-YO embryos displayed a higher intensity of H3K9me3 staining than IVF or NT-YFP embryos (Fig. 5B). H3K27me3 was uniform in all cells at the blastocyst stage (Fig. 6A) and did not differ among treatment groups. The average fluorescence intensity of H3K27me3 was the same among all three groups (Fig. 6B).
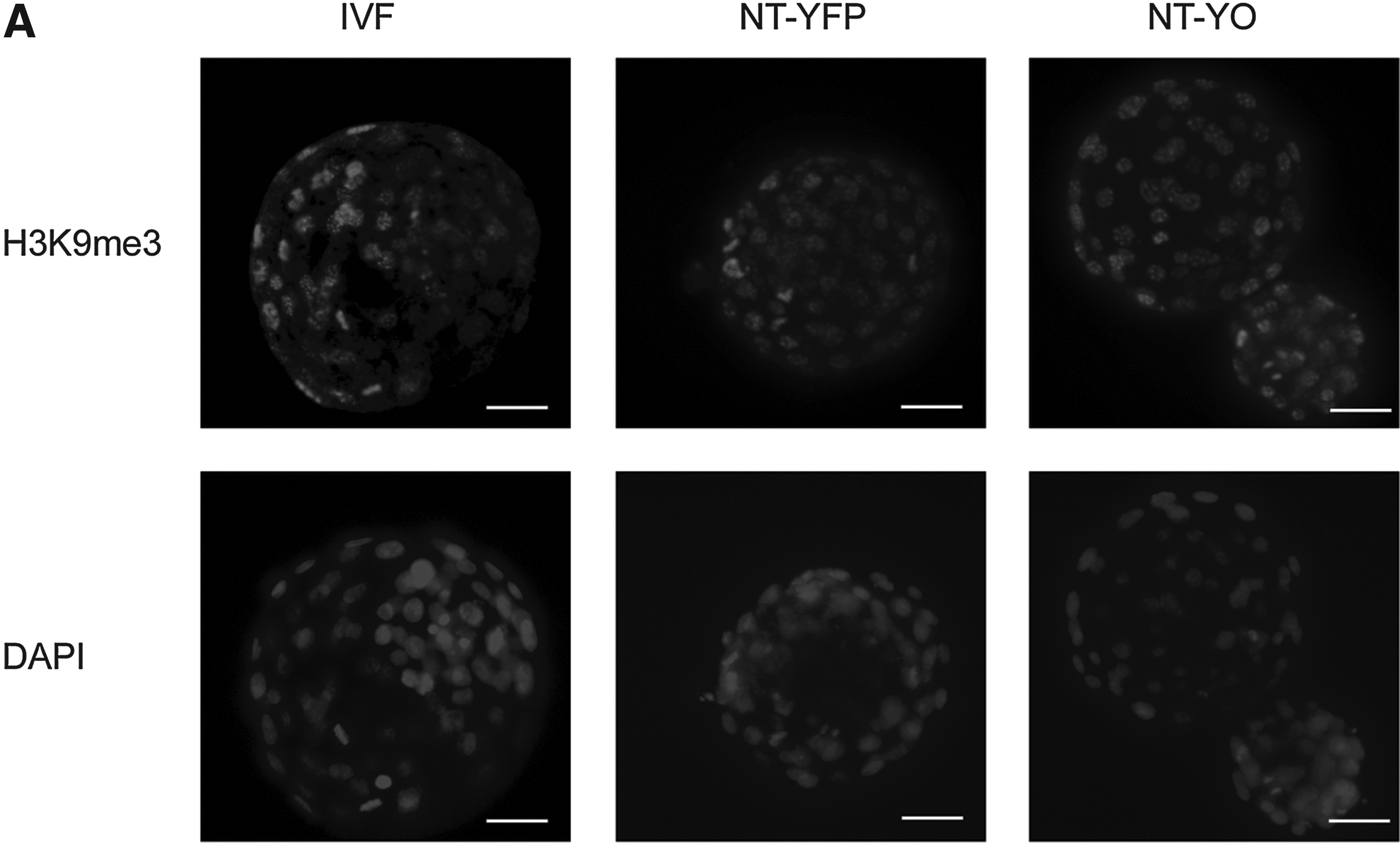
H3K9me3 detection in day 7 blastocysts of IVF, NT-YFP and NT-YO groups. (
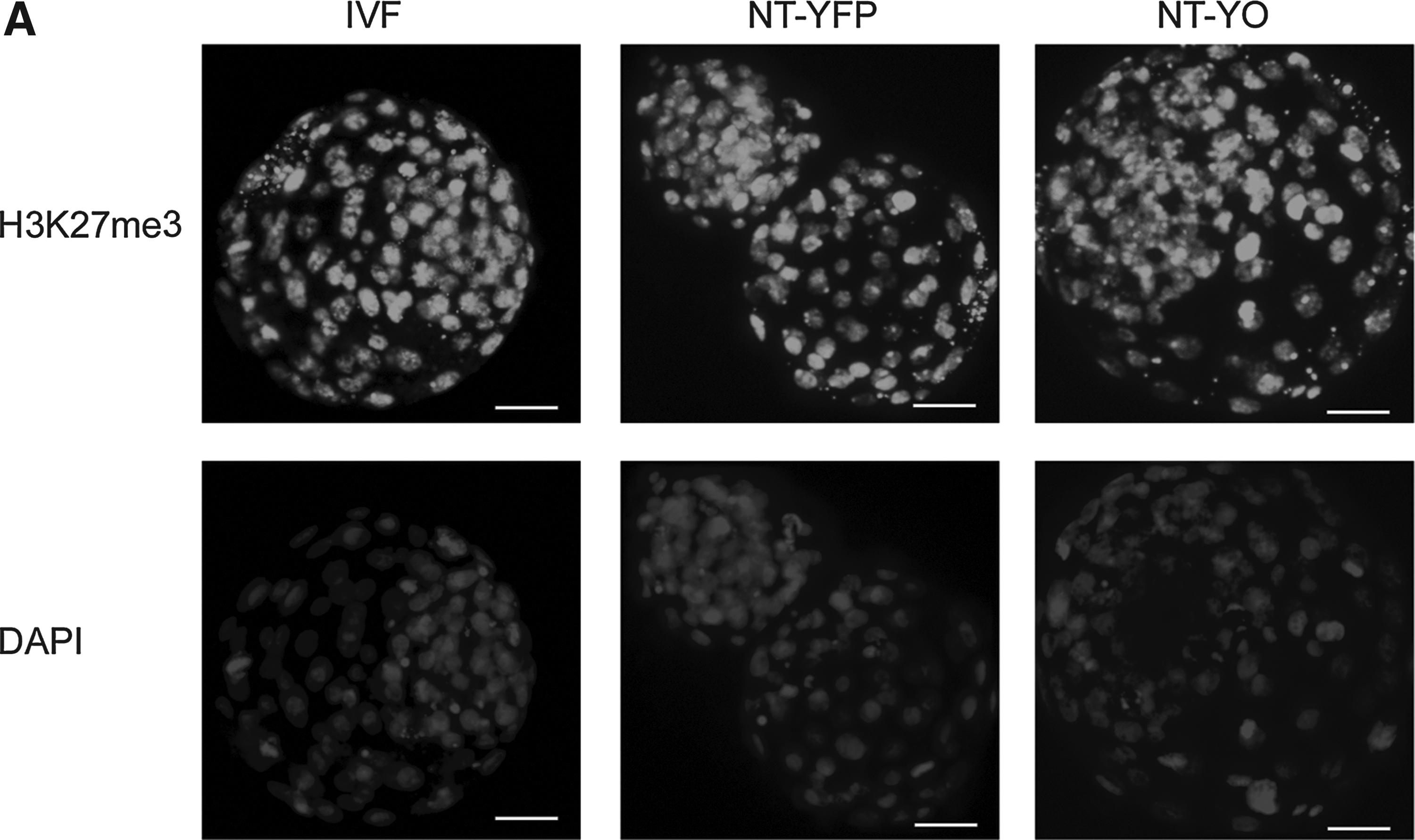
H3K27me3 immunostaining in blastocysts of IVF, NT-YFP, and NT-YO groups. (
Quantitative PCR gene expression analysis
We performed quantitative RT-PCR analysis to verify gene expression changes in pools of IVF, NT-YFP, and NT-YO day-7 blastocysts. We selected genes known for their role during preimplantation development, including genes associated with the H3K9 methylation mark. For EHMT1 and KDM4C, we observed no significant differences in mRNA levels (Fig. 7A). We found that XIST, recently shown to be upregulated in SCNT bovine embryos (Inoue et al., 2010), was upregulated in both the NT-YFP and NT-YO groups compared to the IVF group (Fig. 7B). We assessed endogenous OCT4 expression using primers for the 5′-untranslated region (5′-UTR), and we observed a downregulation in the NT-YFP and NT-YO groups compared to the IVF group (Fig. 7B). Expression of other important genes, such as CDX2, SOX2, and NANOG did not differ among the three groups (Fig. 7B).
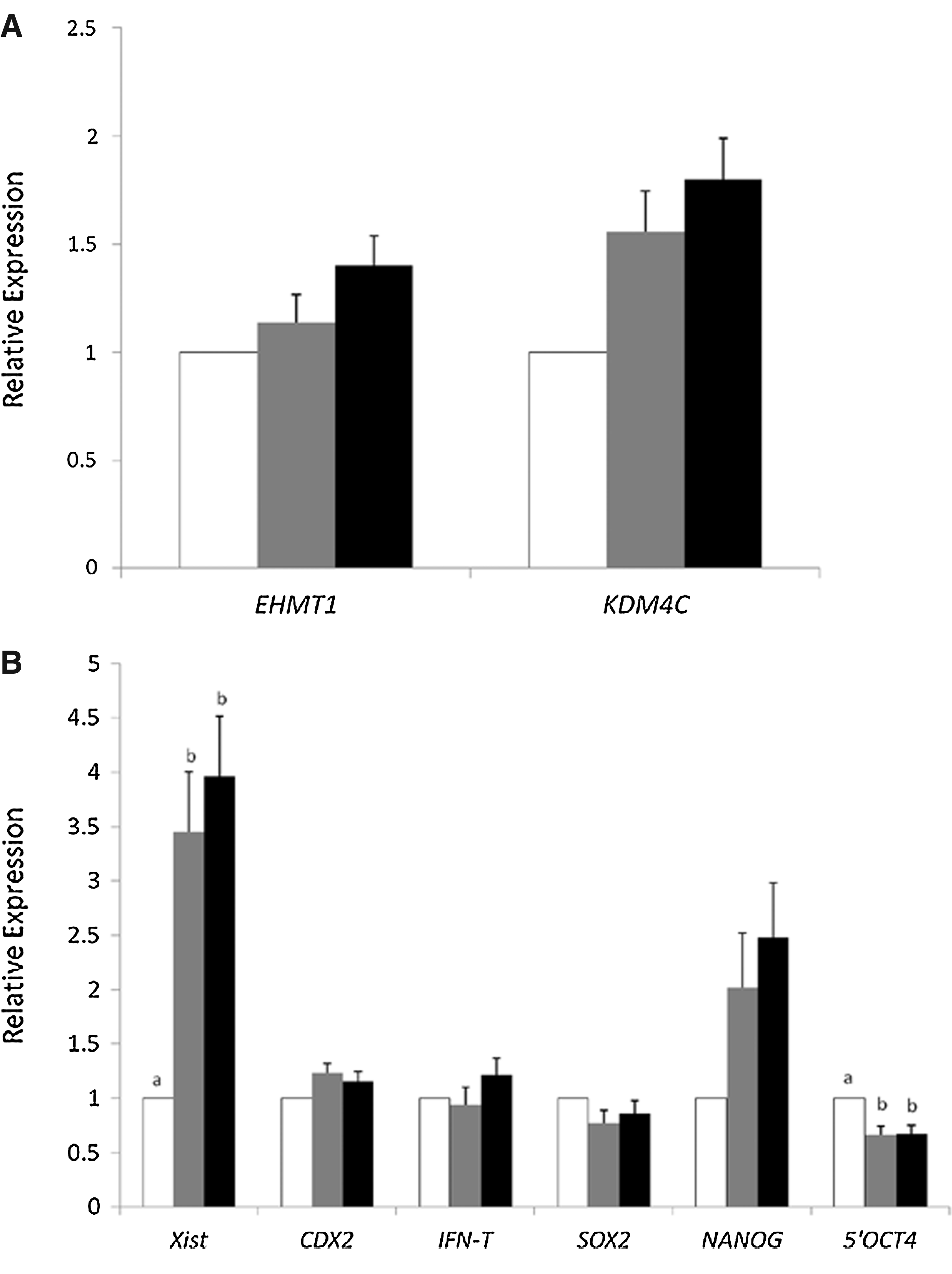
Gene expression analysis. (
Discussion
The SCNT technique has proven itself capable of producing viable cloned offspring; however, its efficiency remains very low. Problems, including low pregnancy rates and high pregnancy losses, are thought to be related to failure in the epigenetic reprogramming of the somatic donor nucleus (Campbell et al., 2007). The use of ESCs in mouse nuclear transfer yields better results, as measured by live offspring, than the use of somatic cells (Rideout et al., 2001), arguing in favor of the notion that a pluripotent nucleus is more easily reprogrammed. OCT4 maintains pluripotency in ESCs (Pesce and Scholer, 2001) and keeps an open chromatin state in embryos, as measured by electron spectroscopic imaging (Ahmed et al., 2010). We hypothesized that we could facilitate the oocyte's task of reprogramming a somatic cell's chromatin by expressing OCT4 in the donor fibroblast prior to SCNT. To test our hypothesis, we generated donor fibroblasts that ectopically expressed YFP and the OCT4-YFP fusion protein. We used immunohistochemistry to confirm proper nuclear localization of OCT4.
Before using these fibroblasts as donor cells, we wanted to determine whether OCT4 overexpression could induce changes in the epigenome. We observed a decrease in H3K9me3 and H3K27me3 after quantification of fluorescence immunocytochemistry. To understand how these changes occurred, we verified the gene expression of the histone methylation enzyme EHMT1 and the demethylation enzymes JARID2 and KDM4C, which promoter regions can be directly bound by OCT4 (Loh et al., 2006). We found the expression of KDM4C, a demethylase that acts specifically on the H3K9me3 residue (Whetstine et al., 2006), was higher in YO than in YFP fibroblasts, which might explain the reduction in global methylation levels of this epigenetic mark. H3K27me3 is catalyzed by the polycomb repressive complex 2 (PRC2), and JARID2 is involved in PRC2 recruitment (Li et al., 2010). However, JARID2 is unchanged in YO fibroblasts, strongly suggesting that the reduction of H3K27me3 as a consequence of OCT4 expression in fibroblasts occurs due to a JARID2-independent mechanism and deserves further characterization.
Expression of OCT4 in fibroblasts prior to fusing them to MII oocytes did not affect SCNT cleavage rates or blastocyst formation. The expression of the transgene itself in both groups—control and YFP-OCT4—was absent at 72 h and at the blastocyst stage, the two time points we investigated. The silencing of the transgenes likely occurred due to the use of retroviral vectors that can be actively methylated and silenced during embryo development (Jähner et al., 1982).
SCNT-derived embryos reportedly have lower TE cell numbers than IVF or IVF-derived embryos, and this alteration was thought to play a role in pregnancy losses (Koo et al., 2002). We observed a significant increase in the number of TE cells in embryos reconstructed with BAF overexpressing OCT4. The number of cells in these embryos was similar to IVF ones. Provided that one of the reasons for placental failure in the SCNT embryos is indeed the reduced TE cell numbers at the blastocyst stage, expression of OCT4 in the donor fibroblast should be explored as a strategy to increase pregnancy and birth rates in cloned bovine embryos. The reasons for this increase in TE cell number were not evaluated in our study.
We assessed the levels of histone trimethylation to verify global epigenetic changes in SCNT-derived blastocysts. The H3K9me3 mark has been shown to display an asymmetry at the bovine blastocyst with the ICM showing higher intensity than the TE (Santos et al., 2003). Curiously, we did not observe any asymmetry in our IVF or SCNT embryos. The relatively lower levels of H3K9me3 observed in YO fibroblasts were not maintained in NT-YO blastocysts. In fact, we saw an increase in H3K9me3 compared to IVF and NT-YFP embryos and observed no changes in histone-modifying enzymes directly regulated by OCT4. Other histone methyltransferases, such as EHMT2, ESET, and SUV39H1, might have been upregulated, perhaps as a compensatory mechanism for the reduction in H3K9me3 in donor cells.
We are in the process of characterizing the dynamics of H3K27me3 during bovine preimplantation development and its significance during SCNT. So far, we know that H3K27me3 gradually decreases from the MII oocyte stage up to the morula stage (Ross et al., 2008). The enzyme JMJD3 actively removes methyl groups in a cell cycle–independent manner; soon thereafter, in parallel with embryonic genome activation, a new pattern of H3K27me3 is reestablished (Canovas et al., 2012). This includes the trimethylation of H3K27 of one of the two X chromosomes in its entirety. Despite the fact that the levels of several histone methylation marks are the same in IVF and SCNT bovine blastocysts (Wu et al., 2011), we previously reported differences in H3K27me3 levels depending upon the method of egg activation. Using phospholipase C zeta (PLCz), the protein responsible for oocyte activation at fertilization, causes the methylation levels of SCNT embryos to resemble IVF methylation levels more closely (Ross et al., 2009). Like Wu et al. (2011), we saw no differences in the levels of H3K27me3 among all three groups in the present study. Embryos used in our previous study had a higher number of total cells at the blastocyst stage in all groups compared to the present study, reflecting a slight difference in the developmental stages used in the two studies. We need further research to determine whether H3K27me3 can act as a reliable marker for successful reprogramming. Unfortunately, live tracing of methylation changes is not possible at this time.
Due to the epigenetic alterations we observed in the donor somatic cells when OCT4 was overexpressed, we decided to measure gene expression in SCNT and IVF blastocysts. We saw no difference in SOX2, CDX2, and IFNT expression levels, which agreed with our previous results and those described by others (Fujii et al., 2010; Ross et al., 2009; Wang et al. 2011; Yao et al., 2009). NANOG gene expression was slightly upregulated in SCNT groups versus IVF, as shown previously (Iager et al., 2008). We also assessed the expression of the 5′[UTR region of OCT4 to differentiate from the exogenous open reading frame of human OCT4, and we observed that both SCNT groups had lower expression levels than the IVF group. This result conforms with previous observations (Ross et al., 2009; Wang et al., 2011).
Taken together, these gene expression results underscore the fact that single-gene analysis cannot be used as a marker for predicting successful SCNT reprogramming (Somers et al., 2006). Nonetheless, it was recently shown that XIST expression increased in SCNT-derived mouse male and female embryos (Inoue et al., 2010; Nolen et al., 2005). Moreover, XIST knockout significantly improved development to term in cloned embryos (Inoue et al., 2010). Our results show that both SCNT groups expressed higher levels of XIST than IVF embryos, which might indicate that complete reprogramming was not yet achieved. Nevertheless, it will take more studies to further validate the predictive value of XIST expression in cows as a marker of successful pregnancy outcome.
In summary, we exogenously expressed human OCT4 in bovine donor fibroblasts and generated SCNT-derived blastocysts. Retroviral transfection of OCT4 into adult fibroblasts increased cell division and reduced H3K9 and H3K27 trimethylation levels. However, we appear to have negated our initial hypothesis that exogenous expression of OCT4 would facilitate reprogramming, because embryos were generally not different from control SCNT-derived blastocysts. Both SCNT groups differed in some analyzed end points from IVF-derived embryos, including increased XIST expression and reduced endogenous OCT4 expression. Although our data do not suggest that overexpression of OCT4 will improve overall reprogramming efficiency, it was notable that expression of a single pluripotency factor significantly altered the number of TE cells, moving the SCNT embryos closer to IVF embryos. This suggests that even small changes in histone methylation status and mRNA levels within donor cells might dramatically affect their reprogrammability and the outcome of SCNT. Generation of live offspring is the ultimate proof to test if novel approaches indeed increase the efficiency of SCNT. Unfortunately due to cost related issues we were unable to perform embryo transfers. Overall, we believe that combining OCT4 overexpression with other approaches, such as histone deacetylase inhibitors, might further change the outcome of bovine SCNT.
Footnotes
Acknowledgments
The authors would like to thank Gabriela Saldana for ovary collections and the members of the Cellular Reprogramming Laboratory for support during the execution of this study.
Author Disclosure Statement
The authors declare that they have no competing interests. This work was funded by MSU AgBioresearch and the MSU Foundation. M.D.G. was funded by the Capes Foundation, with a Capes/Fulbright Scholarship, Ministry of Education, Brazil.