
Abstract
Abstract
Human induced pluripotent stem cells (hiPSCs) need to be generated and expanded under clinically applicable culture conditions before they can be used for clinical application. In this study, we demonstrate that inactivated human mesenchymal stem cells (hMSCs) from different donors can be used as feeder cells to support the establishment and maintenance of hiPSCs. The hiPSCs we generated and expanded on hMSCs exhibited the typical morphology of human embryonic stem cells (hESCs), expressed undifferentiated pluripotent cell markers and genes, differentiated into all three germ layers via embryoid body and teratoma formation, and retained a normal chromosomal karyotype after 14 passages. However, we found that the rate of hiPSCs generation on hMSCs was 7.26%±2.09% compared with that on mouse embryonic fibroblasts (MEFs), and the calculated expansion efficiency of hiPSCs on hMSCs was lower than that on MEFs. hMSCs from various donors and different passages did not influence the results. These findings suggest that hMSCs can be used as feeder cells to derive and maintain hiPSCs, and thus provide another clinically feasible method for generating and expanding hiPSCs. However, the cytokines and adhesion molecules in this system should be identified to develop a preferable clinical culture condition for hiPSCs.
Introduction
As human embryonic stem cells (hESCs), hiPSCs are typically derived and propagated through co-culture with primary mouse embryonic fibroblasts (MEFs) as feeder cells. However, the use of animal feeder cells may transfer exogenous antigens and viruses to hiPSCs, which limits their clinical use. Although feeder-free cultures of hESCs and hiPSCs have been developed (Li et al., 2005; Ludwig et al., 2006; Nagaoka et al., 2010), they may be a hostile environment and are more prone to lead to karyotypic abnormalities in hESCs and hiPSCs (Catalina et al., 2008; Imreh et al., 2006). Consequently, we focused on the use of suitable human feeder cells for pluripotent stem cell propagation.
Previous reports have shown that some kinds of human cells can be used to support hESCs propagation (Hovatta et al., 2003; Lee et al., 2005; Richards et al., 2002). Human mesenchymal stem cells (hMSCs) are multipotent cells that can be isolated from bone marrow (BM), adipose tissue, umbilical cord blood, the umbilical cord, placenta, muscle, liver, and so on, and can replicate as undifferentiated cells in vitro (Nauta and Fibbe, 2007; Pittenger et al., 1999). Studies have shown that culture-expanded hMSCs fully support prolonged hESCs expansion in cultures; these expanded hESCs maintained their pluripotency and normal diploid karyotype (Cheng et al., 2003). Thus, we determined whether hMSCs could be used as feeder layers to support the derivation and propagation of hiPSCs. In this study, we show that culture-expanded hMSCs derived from human adult BM can replace MEFs to support the generation and propagation of hiPSCs. After more than 14 proliferation passages, the hiPSCs retained their undifferentiated state and normal karyotype. The generation efficiency and growth kinetics of hiPSCs on hMSC feeder cells were different from that on MEFs.
Materials and Methods
Cell culture
Human foreskin fibroblasts were isolated from the foreskins of children after circumcision with the informed consent of the donors' parents. The cells were cultured in Eagle's minimum essential medium (Eagle's MEM; Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Invitrogen). BM samples were aspirated from healthy and consenting adult donors. The hMSCs were isolated from BM according to a previously published method (Pittenger et al., 1999). The hMSCs were grown Dulbecco's modified Eagle medium (DMEM) with low glucose (Invitrogen) and 10% FBS. The fibroblasts and hMSCs were subcultured by treatment with 0.05% trypsin/0.53 mM EDTA (Invitrogen). The number of passages increased with each cycle of trypsin-EDTA treatment and replating. H1 cells were obtained from the National Stem Cell Bank and were grown in hESC medium containing DMEM/F12 (Invitrogen) supplemented with 20% knockout serum replacement (Invitrogen), 0.1 mM nonessential amino acids (NEAA; Invitrogen), 1 mM
Retrovirus production, infection, and iPSCs generation
Moloney-based retroviral vectors (pMXs) that contain the human genes encoding the four cell-reprogramming factors Oct4, Sox2, Klf4, and c-Myc were obtained from Addgene. Each plasmid was co-transfected into the 293T cells with packaging plasmids pCMV-gp and pCMV-G (kindly provided by Professor Jing-Kuan Yee, City of Hope) via calcium phosphate co-precipitation (Peng et al., 2001). Viral supernatants were harvested 48 h after transfection. The human foreskin fibroblasts were infected twice in 2 days with the four retroviral vectors. The day the fibroblasts were infected for the first time was designated as day 1. On day 6, the infected fibroblasts were harvested and plated onto inactivated feeder layers (Takahashi et al., 2007). On day 7, the medium was replaced with hESC medium. Valproic acid (at a working concentration of 2 mM; Sigma) (Huangfu et al., 2008) and vitamin C (at a working concentration of 25 μg/mL, Sigma) (Esteban et al., 2010) were added to enhance the cell reprogramming efficiency. Independent, well-defined colonies were picked 3–4 weeks after infection and expanded on inactivated feeder layers in hESC medium.
RNA isolation, reverse transcription, and PCR
Total RNA was isolated with TRIzol reagent (Invitrogen). Total RNA (0.5 μg) was used for cDNA synthesis, and 3.5 μL of the 20 μL reverse transcriptase (RT) product was amplified using a PrimeScript™ RT-PCR Kit (Takara). Most of the primer sequences used in our study were similar to that of Takahashi et al. (2007), except for troponin T type 2 (TNNT2) (5′-TCATGCCCAA CTTGGTGCCTCCC-3′, 5′-ATTTCCAGCGCCCGGTGACTTTA-3′); alpha-1-antitrypsin (AAT) (5′-GACCAAGGCTGACACTCACGATGA-3′, 5′-CCCAGTTGACCCAGGACG CTCTT-3′); and mammalian achaete-scute homolog 2 (Mash 2) (5′-GCCAGC AAGAAGCTGAGCAAGGTGGAGA-3′, 5′-GACGAGTAGGCGGAACGCGGGGAG-3′).
Alkaline phosphatase staining and immunocytochemistry
Alkaline phosphatase (AP) staining was performed using a Stemgent Alkaline Phosphatase Staining Kit (Stemgent). For immunocytochemistry, the cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing with phosphate-buffered saline (PBS), the cells were treated with PBS containing 5% goat serum and 0.3% Triton X-100 for 45 min at room temperature. The cells were stained overnight using primary antibodies at appropriate dilutions at 4°C. The cells were then stained for 2 h at room temperature with secondary antibodies at the appropriate dilution. The nuclei were stained with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI).
Embryoid body–mediated differentiation
For spontaneous differentiation through embryoid body (EB) formation, the hESCs and hiPSCs were harvested by treating the cells with collagenase IV. Cell clumps were transferred onto low-attachment six-well plates in hESC medium without bFGF. After 8 days as floating cultures with the medium changed every other day, the EBs were transferred onto gelatin-coated plates and cultured in the same medium for additional 8 days. Differentiated cells were harvested for total RNA purification and staining for with markers for the three germ layers.
Teratoma formation
The hESCs and hiPSCs were harvested by trypsin treatment, collected, centrifuged, and resuspended in DMEM/F12 medium. Approximately 2×106 cells to 4×106 cells were injected subcutaneously into nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. At 8 weeks after injection, the tumors were dissected and fixed with 4% paraformaldehyde. The tissues were embedded in paraffin, sectioned, and stained with Hematoxylin & Eosin. The histological examination was performed by a pathologist.
Karyotyping
Karyotyping analyses of hiPSCs were carried out by the Institute of Hematology at the First Affiliated Hospital, Zhejiang University School of Medicine. The cells were incubated in 1/100 volumes of colcemid stock solution (Invitrogen) for 2–3 h, harvested, resuspended in hypotonic solution at room temperature for 15 min, and fixed with 3:1 methanol/glacial acetic acid. After staining, the karyotypes of normal human chromosomes were examined under standard G-banding chromosome analysis.
Statistical analysis
All data are presented as mean±standard error of the mean (SEM). The differences between group means were assessed by analysis of variance (ANOVA) for multiple comparisons using SPSS 16.0. The level of statistical significance was set to p<0.05.
Results
Generation and expansion of hiPSCs on hMSC feeder cells
hMSCs from three donors (2 female donors and 1 male donor, 21–45 years old) at passage 5 were used as feeder cells. The cells were plated onto gelatin-coated dishes at 60–70% confluence and inactivated via 3 h of mitomycin C treatment (10 μg/mL). The protocol used to generate the hiPSCs from foreskin fibroblasts on hMSCs is similar to that used on MEFs. Six days after transduction, the cells were harvested by trypsinization and plated onto inactivated hMSC feeder cells. After approximately 3 weeks, well-defined colonies were picked and cultured continuously on inactivated hMSC feeder cells with hESC medium. The cells formed tightly packed and flat colonies with sharp edges (Fig. 1A). Each cell had a typical hESC morphology characterized by large nuclei and scant cytoplasm (Fig. 1B). We expanded these cells on inactivated hMSCs, with split ratios from 1:2 to 1:3 in each passage.
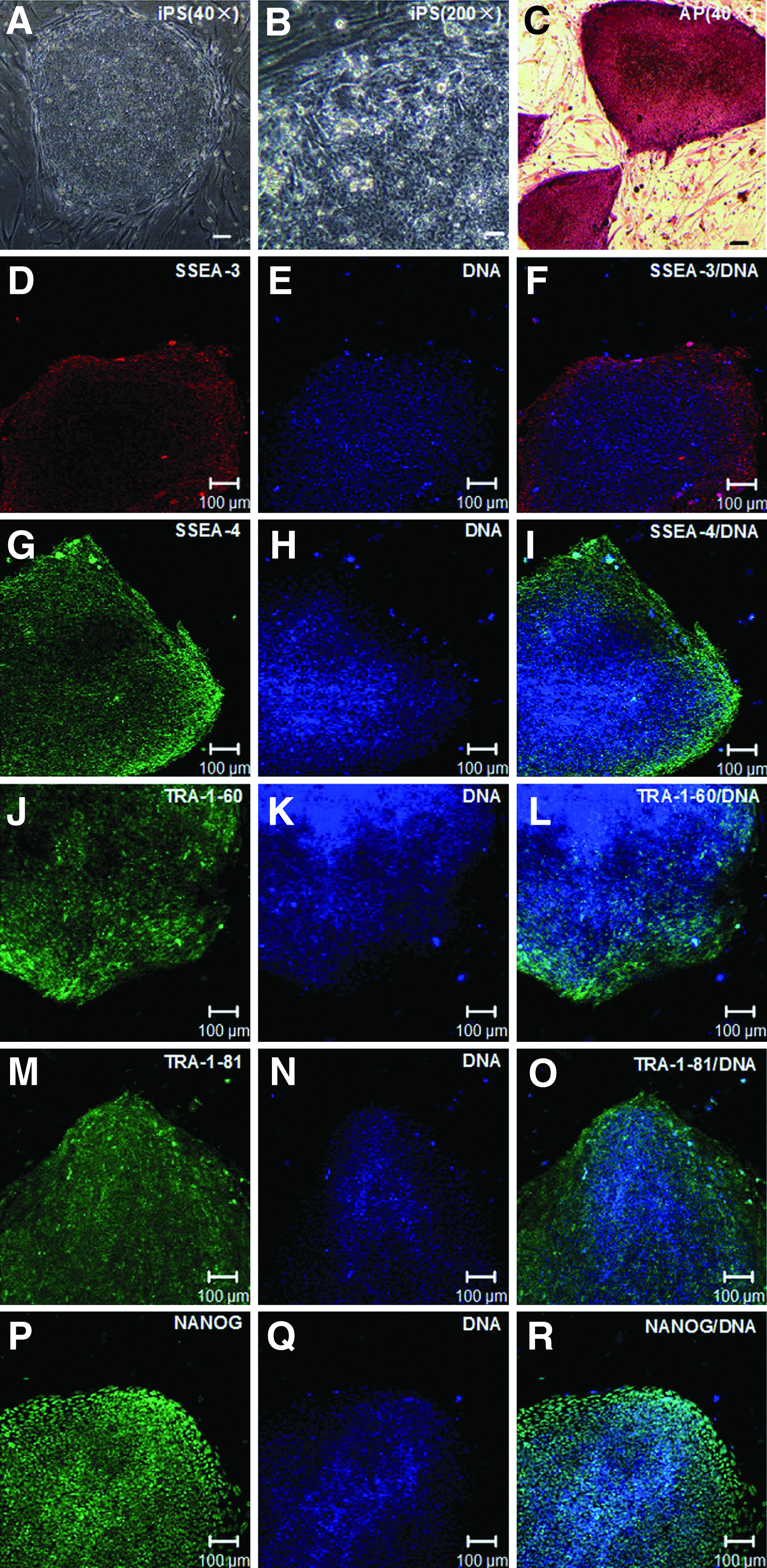
Culture of hiPSCs on hMSCs. (
After 14 passages on hMSC feeder cells, aliquots of the hiPSCs were analyzed for the expression of hESC-specific surface antigens. By AP staining and immunofluorescence analysis, the hiPSC colonies were strongly positive for AP, stage-specific embryonic antigen (SSEA)-3, SSEA-4, tumor-related antigen (TRA)-1-60, TRA-1-81, and NANOG, whereas the hMSC used as feeder cells were almost negative (Fig. 1C–R). By RT-PCR, many undifferentiated hESC marker genes were detected in the hiPSCs cultured on hMSCs (Fig. 2, lanes 3–5). The markers included endo-KLF4, endo-SOX2, endo-cMyc, endo-OCT4, NANOG, DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B), developmental pluripotency-associated 4 (DPPA4), telomerase reverse transcriptase (hTERT), NODAL, reduced expression 1 (REX1), and teratocarcinoma-derived growth factor 1 (TDGF1).
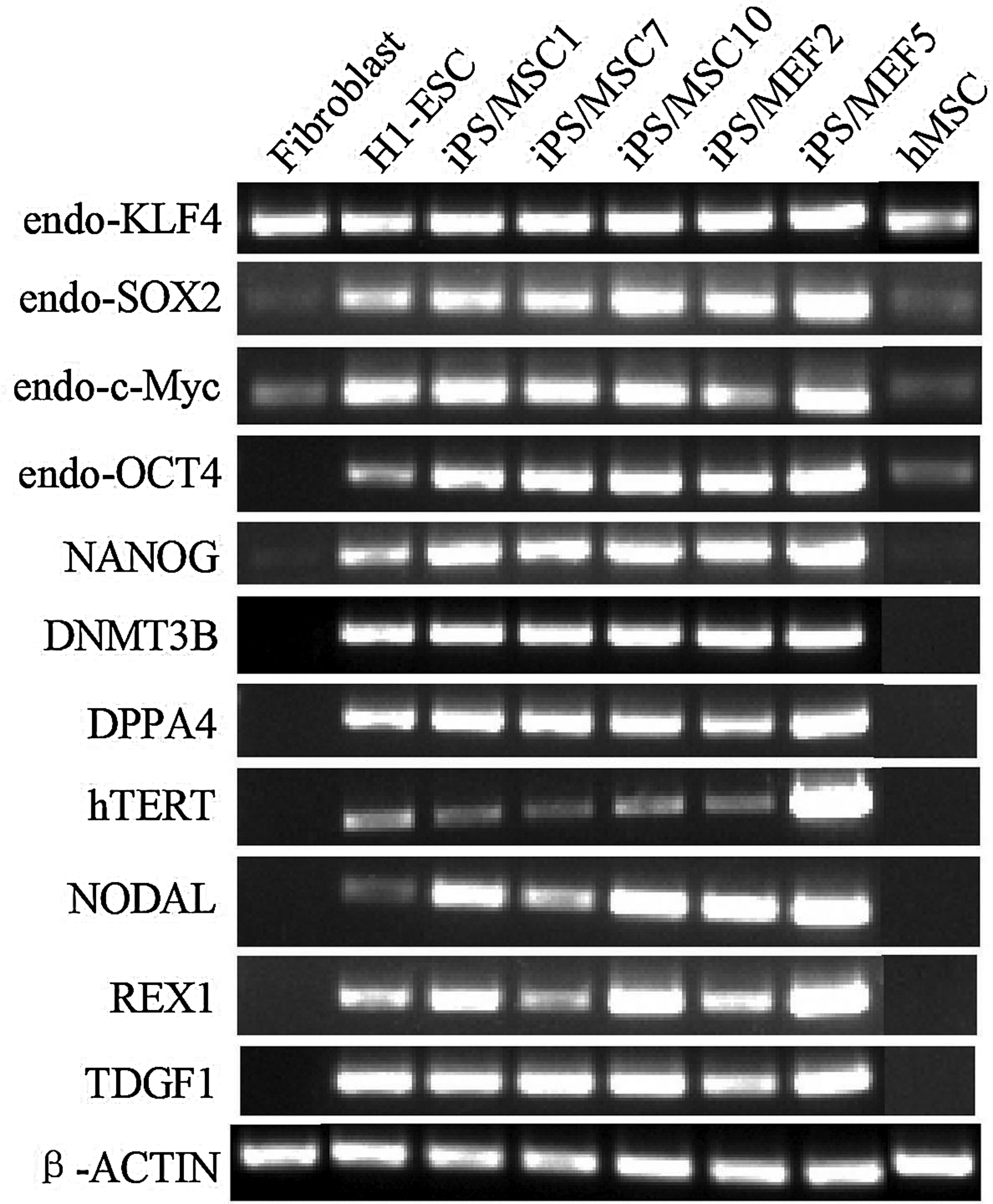
RT-PCR analysis of the expression of hESCs marker genes in hiPSCs cultured on hMSCs. iPS/MSC1,7,10, hiPSCs cultured on hMSC feeder cells; iPS/MEF2,5, hiPSCs cultured on MEF feeder cells. Primers used for oct3/4, sox2, klf4, and c-myc detect only the transcripts of the endogenous genes, not the transgenes.
Expansion hiPSCs on hMSC feeder cells retained pluripotency unique to hESCs
To examine whether the hiPSCs derived and propagated on hMSC feeder cells maintained their unique differentiation potential in vitro and in vivo, we used floating cultivation to form EBs and transplanted these hiPSCs subcutaneously into NOD/SCID mice to form teratomas. Some of two hiPSCs lines (iPS/MSC1, iPS/MSC7) derived and propagated on hMSC feeder cells for 15–16 passages were used.
After 8 days in the suspension culture, the hiPSCs formed round EBs (Fig. 3A). We then transferred the EBs onto gelatin-coated plates and cultivated the EBs for another 8 days. The attached cells derived from the EBs were detected via immunofluorescence analysis. Cells positive for forkhead box A2 (FOXA2, endoderm), myotube (mesoderm), and nestin (ectoderm) were detected in the culture (Fig. 3B–G). In addition, we detected the expression of some differentiation markers for the three germ layers in these differentiated cells through RT-PCT. As shown in Figure 3H, the robust expression of α-fetoprotein (AFP, endoderm), FOXA2 (endoderm), Brachyury (mesoderm), Troponin T type 2 (TNNT2, mesoderm), α-1-antitrypsin (AAT, ectoderm), and mammalian achaete-scute homolog 2 (mash 2, ectoderm) was found in the differentiating cells, whereas OCT4 expression was markedly reduced. These results demonstrated that the hiPSCs derived and propagated on hMSC feeder cells could be differentiated into derivatives of all the three germ layers in vitro.
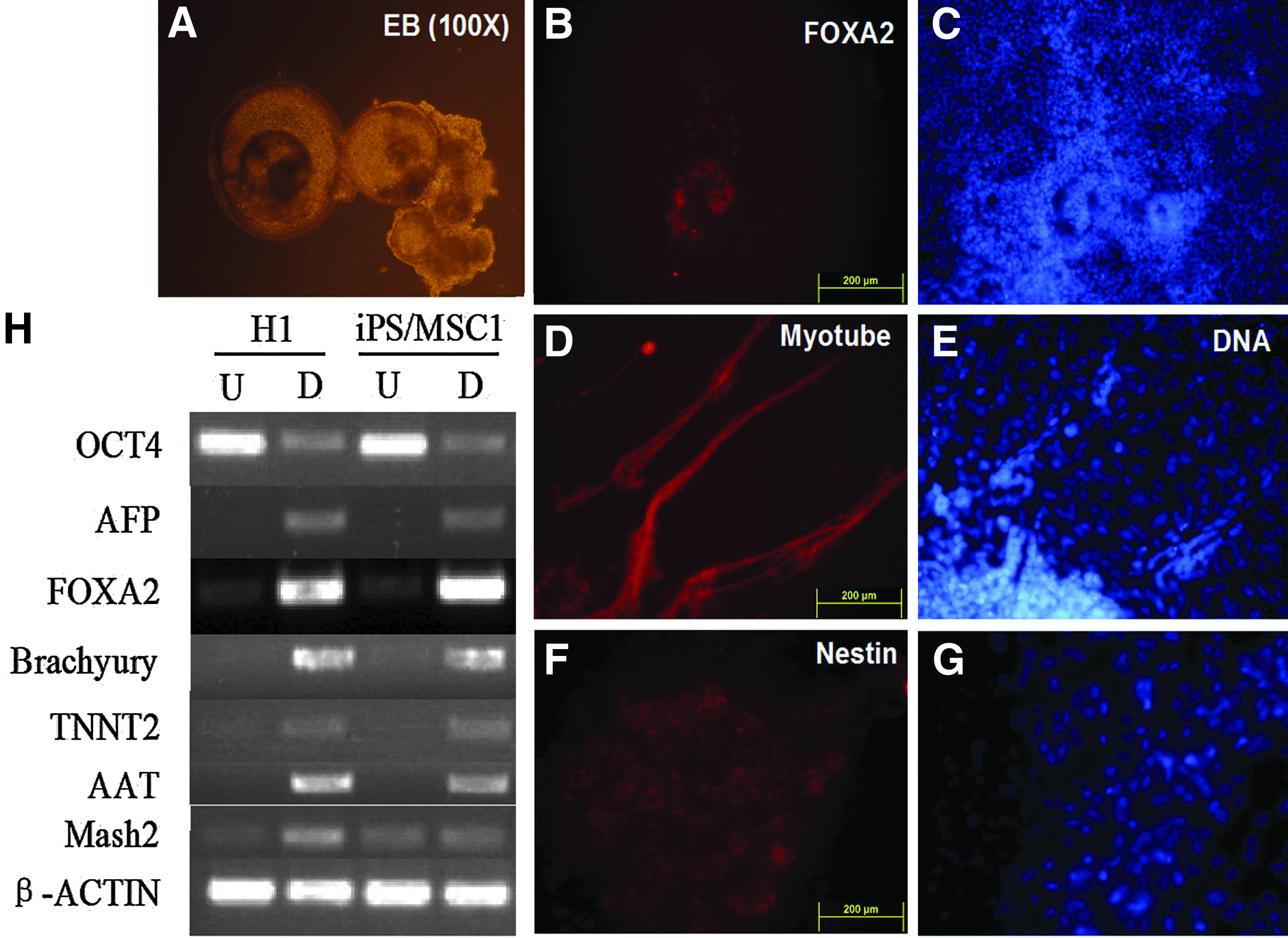
EB-mediated differentiation of hiPSCs cultured on hMSCs. (
To evaluate pluripotency in vivo, we transplanted the hiPSCs subcutaneously into NOD/SCID mice. Teratomas formed about 7–8 weeks after injection (Fig. 4A). Histological study showed that the teratomas contained various tissues that represent the three germ layers, including gut-like epithelial tissues (endoderm) (Fig. 4B, E), cartilage (mesoderm) (Fig. 4C, F), striated muscle (mesoderm) (Fig. 4C, F), and neural tissues (ectoderm) (Fig. 4D, G).
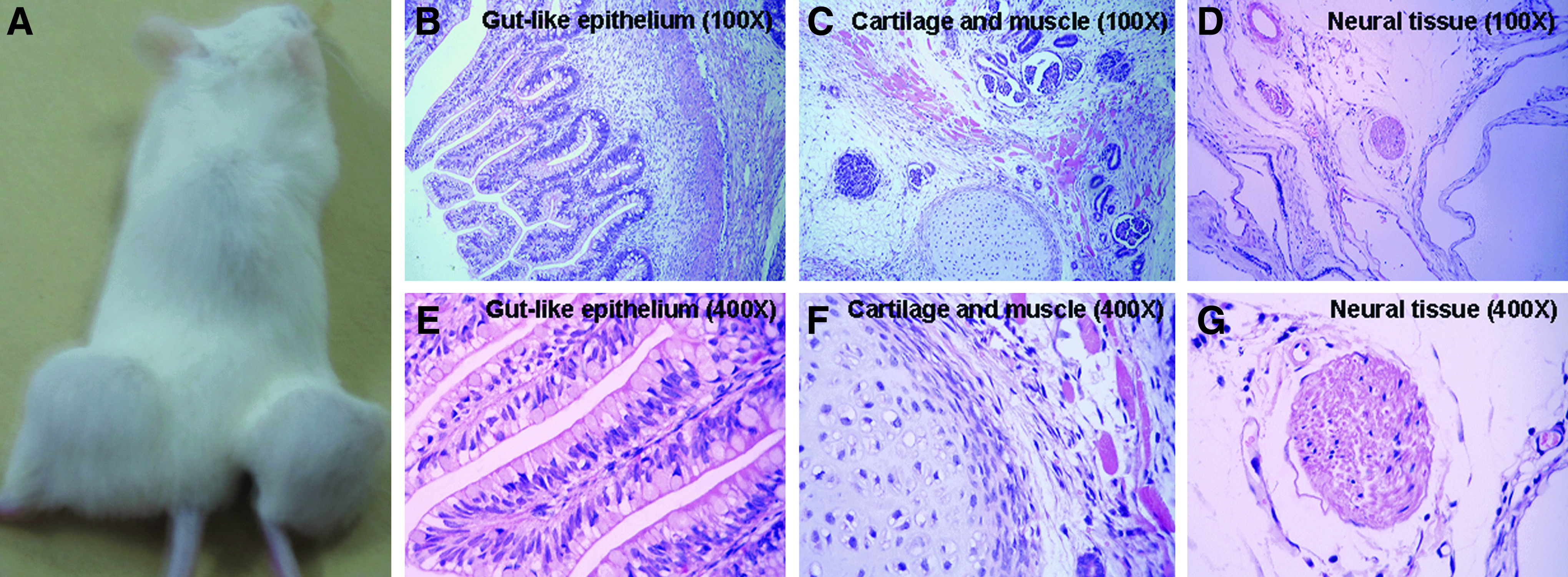
Teratoma formation of hiPSCs cultured on hMSCs (iPS/MSC1 is shown). (
Expansion hiPSCs on hMSC feeder cells retained a normal chromosomal karyotype
We checked the karyotype of the two hiPSCs lines (iPS/MSC1, iPS/MSC7) derived and propagated on hMSC feeder cells after more than 14 passages. To distinguish the hiPSCs (male) from the hMSC feeder cells present in the co-cultures, we propagated aliquots of the hiPSCs lines on hMSC feeder cells from the female donors for more than five passages. Of the two lines examined twice separately, standard G-banding chromosome analysis revealed that both hiPSC lines retained the same normal karyotype (46, XY; Fig. 5), which suggests that expansion hiPSCs on hMSC feeder cells retained a normal chromosomal karyotype.
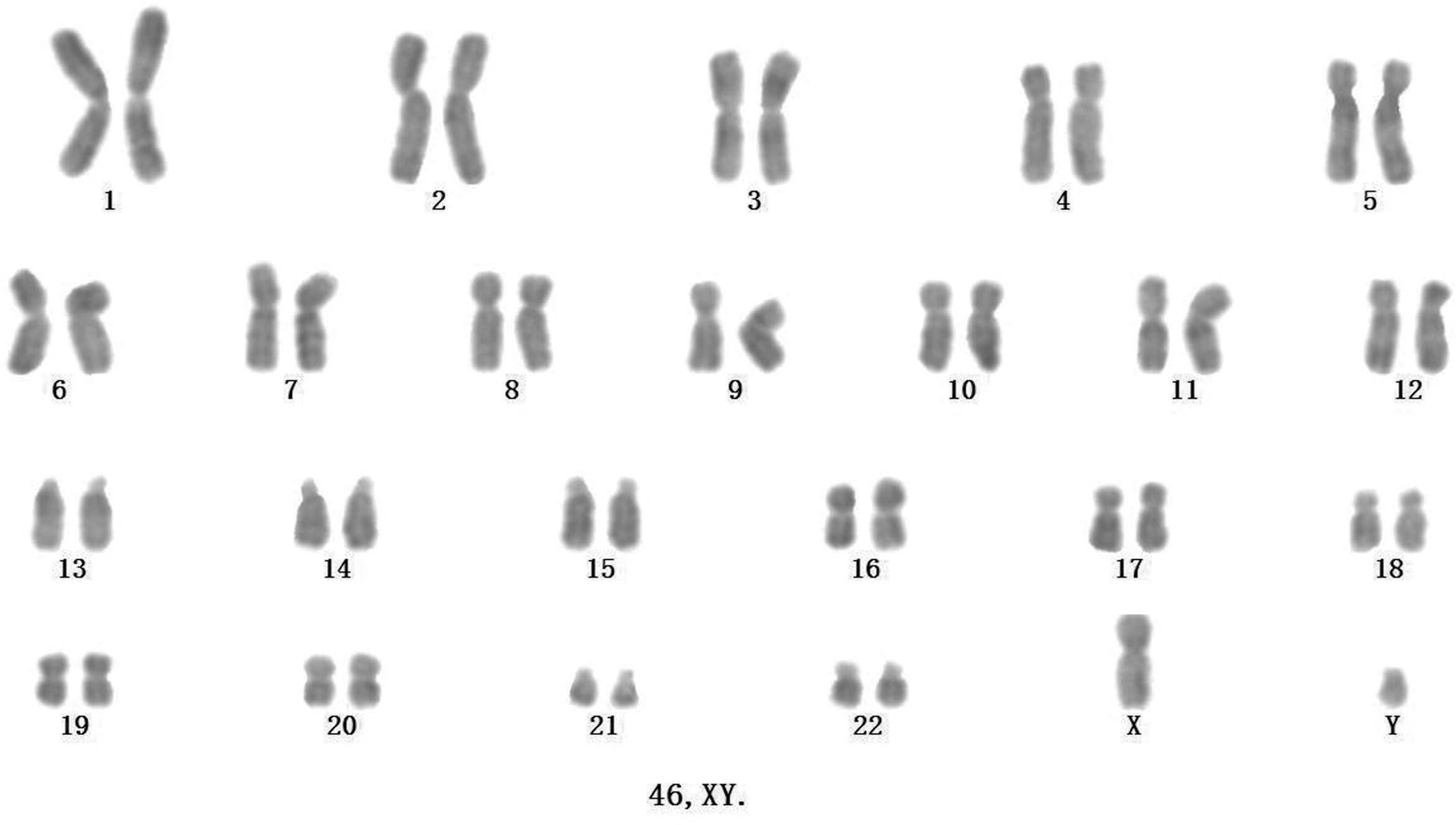
A normal chromosomal karyotype (46, XY) of hiPSCs cultured on hMSCs for more than 14 passages.
Generation efficiency and growth kinetics of hiPSCs on hMSC feeder cells
To compare the generation efficiency of hiPSCs on hMSC feeder cells directly with that on MEF feeder cells, all of the obtained well-defined, hESC-like colonies were counted on the two types of feeder layers. After three independent experiments, the rate of hiPSC generation on hMSC feeder cells was 7.26%±2.09% compared to that on MEF feeder cells (the number of colonies on the hMSC feeder cells divided by the number of colonies on the MEF feeder cells) (p<0.05) (Fig. 6A).
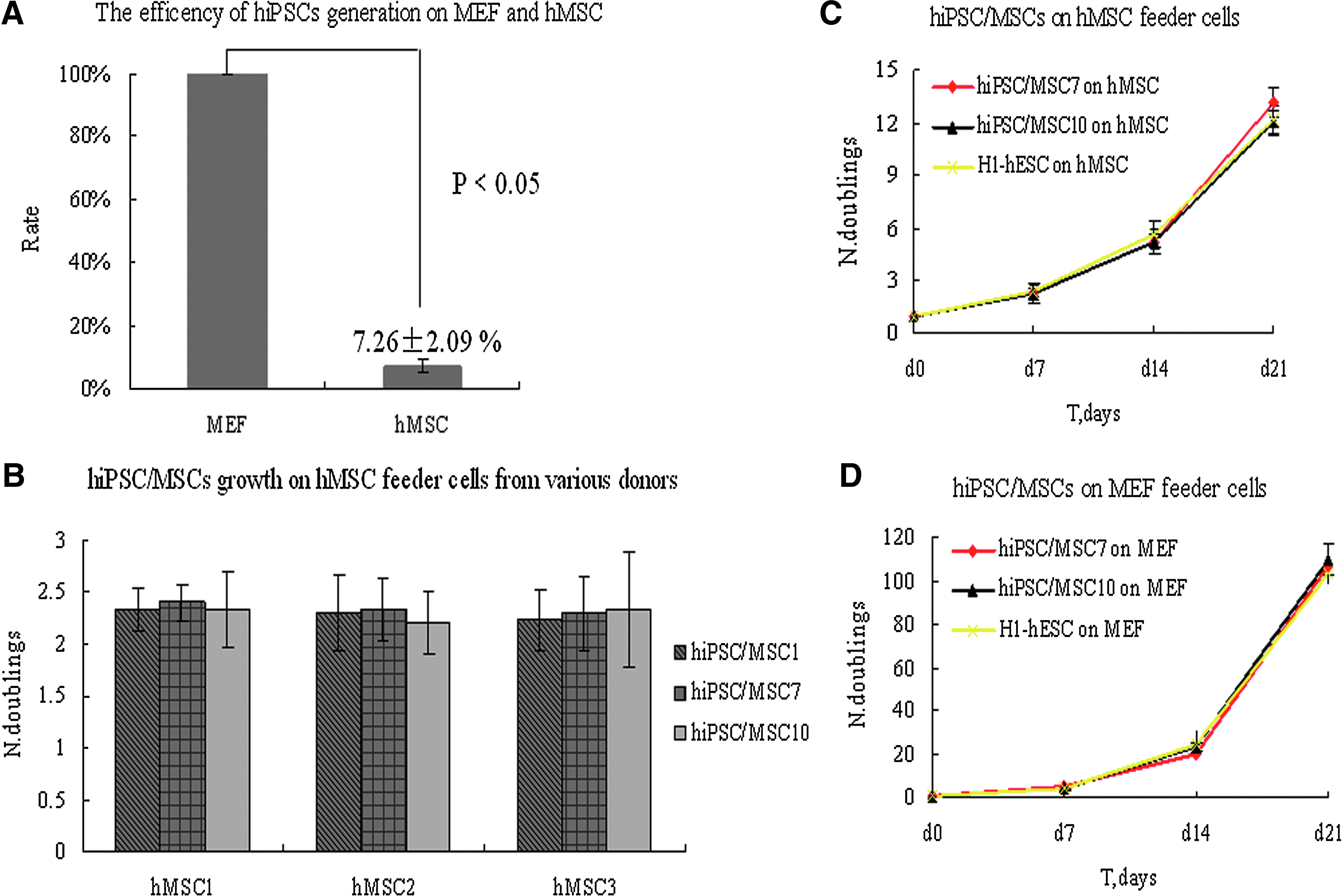
Generation efficiency and growth kinetics of hiPSCs on hMSC feeder cells.
We also detected the growth curve of the hiPSCs maintained on hMSC feeder cells. Some of the P8 hiPSCs lines (iPS/MSC7, iPS/MSC10) were seeded onto tripartite wells of inactivated hMSC and MEF feeder cells. After 7 days, the clones were dissociated with collagenase IV, aliquots of the cells were plated in clumps on newly prepared feeder cells, and aliquots of the cells were made into single-cell suspensions and counted. The average expansion efficiency of three successive passages was calculated, and three independent duplicate experiments were performed. The calculated expansion efficiencies of the hiPSCs on hMSC feeder cells were 2.36±0.14 (clone hiPSC/MSC7) and 2.29±0.03 (clone hiPSC/MSC10) (Fig. 6C). On MEF feeder cells, the expansion efficiencies were 4.89±0.16 (clone hiPSC/MSC7) and 4.49±0.63 (clone hiPSC/MSC10) (p<0.05) (Fig. 6D). The calculated expansion efficiencies of H1-hESC as the control were 2.30±0.13 and 5.01±0.52 on hMSC and MEF feeder cells, respectively (p<0.05) (Fig. 6C, D). The result suggested that the different expansion efficiencies on the different kind of feeder cells were unrelated to the hiPSCs, but were related to the type of feeder cells. The expansion efficiencies of the hiPSCs on hMSC feeder cells from each of the various donors were similar (Fig. 6B).
Discussion
hiPSCs should be generated and expanded under clinically applicable culture conditions before they can be used clinically. Our results first show that inactivated hMSCs from different donors could support the generation and expansion of hiPSCs. The hiPSCs generated and expanded on hMSC feeder cells exhibited morphology typical of hESCs, expressed undifferentiated hESC molecular markers, displayed in vitro and in vivo multilineage developmental potential unique to hESCs, and maintained a normal chromosomal karyotype. Autologous fibroblasts and immortalized human skin fibroblasts have been reported and could be used as feeder layers to establish and maintain hiPSCs (Takahashi et al., 2009; Unger et al., 2009). The hMSCs could be readily derived from healthy adult donors and expanded 1 million–fold before use as feeder cells; the unrelated hMSCs did not generate alloreactive T lymphocytes in vitro and did not display normal alloresponses in vivo (Nauta and Fibbe, 2007). This novel animal cell–free culture system provides another clinically feasible method for generating and expanding hiPSCs.
Our data demonstrate that the generation efficiency of hiPSCs on hMSC feeder cells was lower than that on MEF feeder cells. However, Takahashi et al. (2009) and Unger et al. (2009) did not mention the generation efficiency of hiPSCs on their human feeder cell culture system. Many studies revealed that reprogramming is a multistep process regulated by many factors and signaling molecules. The addition of some molecules and activation or inhibition of these signaling pathways could boost the efficiency of somatic cells reprogramming (Chen et al., 2011; Esteban et al., 2010; Huangfu et al., 2008; Ichida et al., 2009; Li et al., 2009; Lin et al., 2009; Maherali and Hochedlinger, 2009). Proteomic analysis of the secretions of human and mouse feeder cells revealed that the proteins secreted by human and mouse feeder cells differ (Prowse et al., 2007). These different types of proteins involve the WNT, bone morphogenetic protein (BMP), and transforming growth factor-β (TGF-β) pathways, which reportedly regulate the reprogramming process (Chen et al., 2011; Ichida et al., 2009; Li et al., 2009; Maherali and Hochedlinger, 2009). Another study indicated that TGF-β is secreted by hMSCs, and its production increases in response to bFGF (Montes et al., 2009). TGF-β has been described as a negative regulator of reprogramming, at least in part by preventing the mesenchymal-to-epithelial transition (Li et al., 2010). Currently, we have not identified the molecules released by hMSCs to regulate reprogramming. To this end, we will investigate the separation of soluble factors and adhesion molecules generated by hMSCs to determine which of them regulate the reprogramming process and improve the generation efficiency of hiPSCs on hMSC feeder cells.
In our study, the expansion efficiency of hiPSCs on hMSC feeder cells was lower than that on MEF feeder cells. Initially, we suspected that this lower expansion efficiency is due to the characteristics of the hiPSCs derived from our culture system. We compared the expansion efficiency of the hiPSC lines on hMSC feeder cells and those on MEF feeder cells. The hESCs and hiPSC lines derived on the MEF feeder cells were used as the control (the data of the expansion efficiency of hiPSC lines derived on MEF feeder cells are not shown). We found that the expansion efficiencies of the hiPSC lines and the H1-hESC on hMSC feeder cells were similar and were lower than that on MEF feeder cells. The different preparations (passages 3, 5, 7) and different inoculation densities (0.5×104 cells/cm2, 1×104 cells/cm2, 5×104 cells/cm2) of hMSCs were examined, all of which showed similar results (data not shown). To some degree, this result is inconsistent with a previous report by Cheng et al. (2003), which indicated that hESCs grow better on hMSC feeder cells than on MEF feeder cells once they have adapted to the hMSCs. Some signaling pathways, such as the BMP4, TGF-β, FGF4, WNT, and NODAL signaling pathways, were implicated in the maintenance of pluripotent stem cell self-renewal and growth (Brandenberger et al., 2004; Rho et al., 2006). The proliferation of hESCs/hiPSCs in culture was expected to differ among different culture conditions. In our observations, optimizing culture of hESCs/hiPSCs is still best achieved on MEF feeder cells if the potential clinical application of hESCs/hiPSCs is not considered. We should also determine the molecular mechanism of hESC/hiPSC self-renewal and growth and develop a preferable clinical culture condition for hESCs/hiPSCs.
Footnotes
Acknowledgments
This work was supported by National Natural Science Foundation of China (NSFC) grants (81000198), Zhejiang Provincial grants (2009C14011), and Doctoral Program of Higher Education of China grands (20100101110121). We thank Professor Jing-Kuan Yee for the retroviral system. We are also grateful to Zhimei Chen, Youfa Zhu for their excellent technical assistance.
Author Disclosure Statement
The authors declare no financial conflict of interest.