
Abstract
Abstract
Stem cells used for clinical tissue regeneration therapy should have the capacity of self-renewal, high proliferation, and differentiation and be able to be transplanted in large numbers. Although high concentrations of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) may induce the differentiation of stem cells, these factors have been widely used to enhance the propagation of stem cells, including adipose-derived mesenchymal stem cells (ASCs). However, the effects of low concentrations of EGF and bFGF on stem cells need to be evaluated carefully. This study illustrates that low concentrations of EGF (5 ng/mL) and bFGF (10 ng/mL) increase the proliferative ability of ASCs and induce the typical spindle-shaped cell morphology. EGF and bFGF added to medium promoted neural lineage differentiation and impaired the mesodermal differentiation ability of ASCs. This study demonstrates that even low concentrations of EGF and bFGF may limit the differentiation ability of stem cells during stem cell expansion in vitro. EGF and bFGF supplementation should be carefully considered in stem cells for clinical applications.
Introduction
Adipose-derived stem cells (ASCs) may be useful “seed” cells for cellular therapy applications because they are more readily acquired, relatively safe, and easy to expand in vitro (Gimble et al., 2007; Gimble et al., 2011; Lindroos et al., 2011; Majka et al., 2011; Zuk et al., 2001). Previous studies have shown that 5 ng/mL EGF or 10 ng/mL bFGF can promote the expansion of ASCs (Baer et al., 2009; Hauner et al., 1995; Lee et al., 2004; Tapp et al., 2009). However, little is known about the effect of these pro-survival and pro-proliferative concentrations of EGF and bFGF on the differentiation of ASCs.
To investigate whether EGF and bFGF can influence the stemness and differentiative ability of ASCs when enhancing the proliferation of ASCs, we cultured ASCs in medium supplemented with 5 ng/mL EGF and 10 ng/mL bFGF. The results of this study suggest that EGF and bFGF, even at low concentrations, can direct the fate of ASCs toward a neural lineage in vitro, which may limit the usage of stem cells in specific clinical trials.
Materials and Methods
Isolation and culture of ASCs
ASCs were obtained from 4-week-old female Sprague-Dawley rats (weight, 100–130 grams, n=9), as previously described (Zuk et al., 2001). Briefly, inguinal fat tissue was washed with sterile phosphate-buffered saline (PBS) and digested with 0.1% collagenase I in a water bath at 37°C for 1 h, and the cells were centrifuged at 800 rpm for 5 min. The cells were resuspended in either (1) unsupplemented basal medium (UN medium) only composed of Dulbecco's modified Eagle medium (DMEM; Thermo Fisher Scientific, West Sussex, UK), 5% fetal bovine serum (FBS; Thermo Fisher Scientific, West Sussex, UK), and 1% penicillin/streptomycin (Invitrogen Corp., Carlsbad, CA, USA); or (2) EF medium, which consisted of basal medium containing 5 ng/mL EGF (Gibco Lab., NY, USA) and 10 ng/mL bFGF (Peprotech, NJ, USA). The cells were seeded into T25 flasks and cultured at 37°C in 5% CO2. Approximately 1 day after initial plating, the media were removed and replaced with fresh media to remove nonadherent cells. On day 7 (D7), the cells were washed with PBS, incubated in 0.25% trypsin (Gibco Lab., NY, USA) at 37°C for 2 min, and subcultured at 1:3. The cells were routinely subcultured every 3 days thereafter. All experimental procedures were conducted according to Southeast University, Medical Faculty ethic committee approval.
Analysis of ASC morphology, proliferation, and cell cycle distribution
On D20, the morphology of ASCs cultured in UN or EF medium at 90% confluence was assessed by bright-field light microscopy (Zeiss Ti-S Germany, Oberkochen, Germany). Then, the same cells were incubated in 0.25% trypsin and plated into 12-well plates at 1×103 cells/mL; the cell numbers were counted daily for 8 continuous days. The cell numbers were calculated in the log phase of the resulting growth curves. Cell cycle analysis was performed using propidium iodide (PI; Sunshine, Nanjing, China) staining and flow cytometry (Accuri C6; BD, Michigan, USA). The cells were detached, centrifuged, fixed with 70% cold (4°C) ethanol for 0.5 h, and 1 mL of 50 μg/mL PI and 20 μg/mlL RNase A (Sunshine, Nanjing, China) were added. The cells were incubated for 0.5 h in the dark and filtered through a 100-μm nylon mesh to remove clumps. Data analysis was performed using C-Flow software (BD, Michigan, USA) and FlowJo analysis software (FlowJo; Ashland, OR, USA). All experiments were performed in triplicate.
Osteogenic and neural differentiation
On D20, ASCs cultured in UN or EF medium were seeded into 12-wells plate at a density of 1×103 cells/ mL. After 1–2 days, the media were replaced with osteogenic differentiation medium, which was replaced every 3 days for a period of 15 days. The osteogenic differentiation medium was UN or EF medium supplemented with 10 mmol/L glycerol phosphate disodium salt hydrate, 10 nmol/L dexamethasone, 50 μmol/L
After 7 days induction, an alkaline phosphatase (AP) detection kit (Amresco, Solon, OH, USA) with 5-bromo-4-chloro-3-indolyl phosphate/p-nitroblue tetrazolium chloride (BCIP/NBT) as a substrate was used to assess osteogenic differentiation. The cells were fixed with 4% paraformaldehyde for 30 min, rinsed three times with PBS, and BCIP/NBT was added. The cells were incubated for 30 min, and the samples were rinsed once with water and observed using bright-field light microscopy. Neural differentiation was also assessed after 15 days induction using immunofluorescent staining and PCR, as described below.
Neural differentiation was performed in a similar manner over a period of 15 days using UN or EF medium supplemented with 100 ng/mL retinoic acid (RA; Sigma-Aldrich, St. Louis, MO, USA). Neural differentiation was assessed after 15 days induction using immunofluorescent staining and PCR, as described below.
Immunofluorescent analysis
Immunofluorescent analysis was employed to detect the expression of stemness markers in cells cultured in UN or EF medium. After 20 days of culture, the cells were fixed with 0.5% formaldehyde containing 0.2% Triton X-100 in PBS buffer at pH 7.4 for 5 min at room temperature, rinsed once with PBS, fixed again in 4% formaldehyde in PBS for 20 min, and rinsed three times with PBS. The samples were incubated with rabbit anti-rat primary antibodies against the stemness markers Oct4 and Sox2 (1:100, Santa Cruz Biotechnology, CA, USA) in 1% bovine serum albumin (BSA; Sunshine, Nanjing, China) at 4°C for at least 12 h.
After 15 days of induction in UN and EF osteogenic differentiation medium, ASCs were subjected to immunofluorescent staining using rabbit anti rat osteocalcin (Ocn) primary antibody (1:100; in 1% BSA; Santa Cruz Biotechnology, CA, USA) to detect differentiated osteogenic cells. After 15 days induction in UN and EF neural differentiation media, ASCs were subjected to immunofluorescent staining using rabbit anti-rat nestin and glial fibrillary acidic protein (Gfap) primary antibodies (1:100 in 1% BSA, Santa Cruz Biotechnology, CA, USA) to detect differentiated neural cells.
After washing with PBS, the samples were incubated with the appropriate secondary antibody (1:200 in 1% BSA; goat anti-rabbit Alexa-Fluor 488 or donkey anti-rabbit Alexa-Fluor 647; Invitrogen Corp., Carlsbad, California, USA) for 1 h at 37°C while protected from light. Then, the samples were washed twice with PBS, the nuclei were detected by staining with 10 μg/mL Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) for 30 min while protected from light, and images were obtained using a Revolution XD confocal laser scanning microscope (Andor, Belfast, Northern Ireland).
RNA isolation, reverse transcription polymerase chain reaction, and real-time quantitative fluorescence PCR
ASCs cultured in EF or UN medium were harvested on D10, D20, and D30, and the expression of Oct4, Sox2, Klf4, Nanog, and Lin28a was measured to assess the changes in ASC stemness markers. After induction with osteogenic medium for 15 days, the expression of Alp, Ocn, core binding factor alpha (Cbfa), and collagen type I (Col) was quantified to assess osteogenic differentiation. After induction with neural medium for 15 days, the expression of Nestin, Gfap, and microtubule-associated protein-2 (Map2) was quantified to assess neural differentiation. Glycerol-3-phosphate dehydrogenase (Gapdh) was used as a control.
Total cellular RNA was isolated using the TRIzol method (Invitrogen Corp., Carlsbad, California, USA). DNase-treated total RNA (20 μL total volume) was incubated with 1 μL of 50 mM oligo(dT18) (TaKaRa, Dalian, China). After denaturation, 5× buffer, dNTPs, RNase inhibitor, and PrimeScript Reverse Transcriptase (TaKaRa, Dalian, China) were added, as specified by the manufacturer's protocol. Reverse transcription was performed for 60 min at 42°C, followed by 5 min at 85°C to inactivate the reverse transcriptase.
Reverse transcription polymerase chain reaction (RT-PCR), and real-time quantitative fluorescence PCR (qRT-PCR) were performed using standard protocols. Specific primers were designed for each gene based on the GenBank sequences; the primer sequences are listed in Table 1. RT-PCR was carried out at 95°C for 1 min, 95°C for 15 sec, 57°C/60°C for 40 sec, and 72°C for 40 sec (28–37 cycles). qRT-PCR was performed on the Applied Biosystems 7500 Sequence Detection System (BD, Michigan, USA). Briefly, 1 μL cDNA was added to 10 μL of 2× SYBR Green PCR master mix (TaKaRa, Dalian, China) and 200 nM of each primer in a total volume of 20 μL. The reactions were amplified over 40 cycles of 95°C for 15 sec and 60°C for 1 min. Afterward, a thermal denaturation protocol was performed to determine the number of products present in each reaction. The reactions were typically run in triplicate. The cycle number at which the reaction crossed an arbitrarily placed threshold (Ct) was determined for each gene. Target gene expression was normalized to the expression of Gapdh in each sample. Data were analyzed using the 2−ΔΔCt method(Livak et al., 2001).
Statistical analysis
All data were expressed as the mean &plumn; standard deviation (SD). Differences were compared using the Student t-test; p values <0.05 were considered statistically significant (*p<0.05, **p<0.01).
Results
EGF and bFGF enhance the proliferation of ASCs
ASCs were isolated from adipose tissue, according to a previously published protocol, and the influence of EFG and bFGF on cell morphology, proliferation, and cell cycle distribution was analyzed. ASCs grew in colonies and continuously expanded until they approached confluency; the cells displayed stable and rapid growth in vitro. The expression of ASC surface markers was confirmed by flow cytometry (data not shown). ASCs cultured in UN medium displayed a heterogeneous morphology; most of the cells had a broad, flattened morphology and extended pseudopods (Fig. 1a). In contrast, ASCs cultured in EF medium displayed evident central nucleoli, their cytoplasm contained elongated and spindle-shaped processes, and short protrusions appeared on the surface of the cell bodies (Fig. 1b).
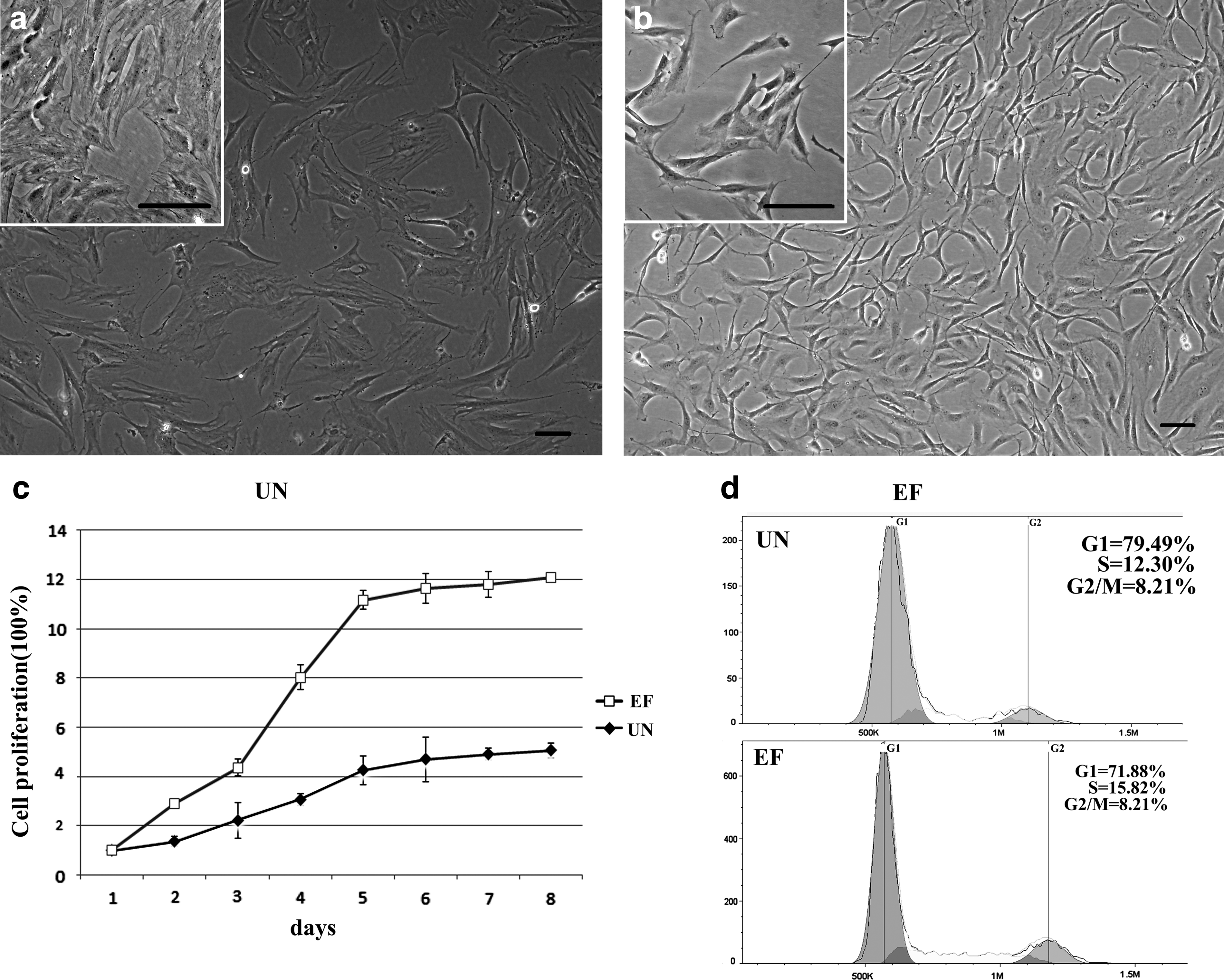
Effects of EGF and bFGF on the morphology, proliferation and cell cycle distribution of ASCs. (
To investigate the biological effects of EGF and bFGF on ASCs, the number of cells was recorded every day for 8 days and cell cycle distribution was analyzed by flow cytometry. ASCs displayed logarithmic growth curves in both UN and EF media. After an initial lag or stationary period, the cells expanded rapidly in a logarithmic manner, until they reached a plateau. However, ASCs grew significantly faster in the presence of EGF and bFGF (Fig. 1c). On day 8, the number of cells in the EF group was three-fold higher than the UN group.
The cell cycle analysis (Fig. 1d, e) indicated that, compared to UN medium, the ratio of G1-phase ASCs in the EF medium decreased from 79.49% to 71.88% (p<0.05), and the ratio of G2/M phase increased from 8.21% to 12.30% (p<0.05). These data demonstrate that EGF and bFGF stimulate the growth of ASCs and enhance their proliferative capacity.
EGF and bFGF alter the pluripotency of ASCs
Alhough EGF and bFGF can stimulate ASC proliferation, the effect of these growth factors on the maintenance of stemness should be carefully investigated. Oct4 and Sox2, markers of undifferentiated stem cells, are critically involved in self-renewal (Driessens et al., 2011; Wang et al., 2012b). Immunofluorescent staining was used to detect the expression of both Oct4 and Sox2 in ASCs cultured in UN and EF media for 20 days (Fig. 2a, b, c, d). The level of Oct4 expression in ASCs cultured in EF medium was lower than in UN medium (Fig. 2 a, b). However, the expression of Sox2 was higher in ASCs cultured in EF medium (Fig. c, d).

EGF and bFGF affect the pluripotency of ASCs. Immunofluorescent staining and bright-field micrographs (BF) of ASCs after 20 days of culture in UN or EF medium. (
RT-PCR and qRT-PCR were used to assess the pluripotency of ASCs cultured in EF medium on D10, D20, and D30. As shown in Figure 2e, the levels of Sox2, Klf4, and Nanog initially increased by D20 and were further upregulated by D30. The expression of Sox2, Klf4, and Nanog increased 3.45-fold, 9.47-fold, and 3.89-fold by D30 compared to D10 (Fig. 2f). In contrast, the expression of Oct4 and Lin28a was downregulated 2.38-fold and 1.76-fold, respectively (Fig. 2f). These results indicate that culture in medium supplemented with 5 ng/mL EGF and 10 ng/mL bFGF may affect the pluripotent state of ASCs.
EGF and bFGF impair the osteogenic differentiation potential of ASCs
The results in this study demonstrated that EGF and bFGF increased the expression of Sox2 and decreased the expression of Oct4 in ASCs, which indicates that EGF and bFGF may induce ASCs to undergo neural lineage differentiation and impair their ability to undergo osteogenic differentiation. To investigate this, osteogenic differentiation was induced by culturing ASCs in UN or EF medium containing glycerol phosphate disodium salt hydrate, dexamethasone,
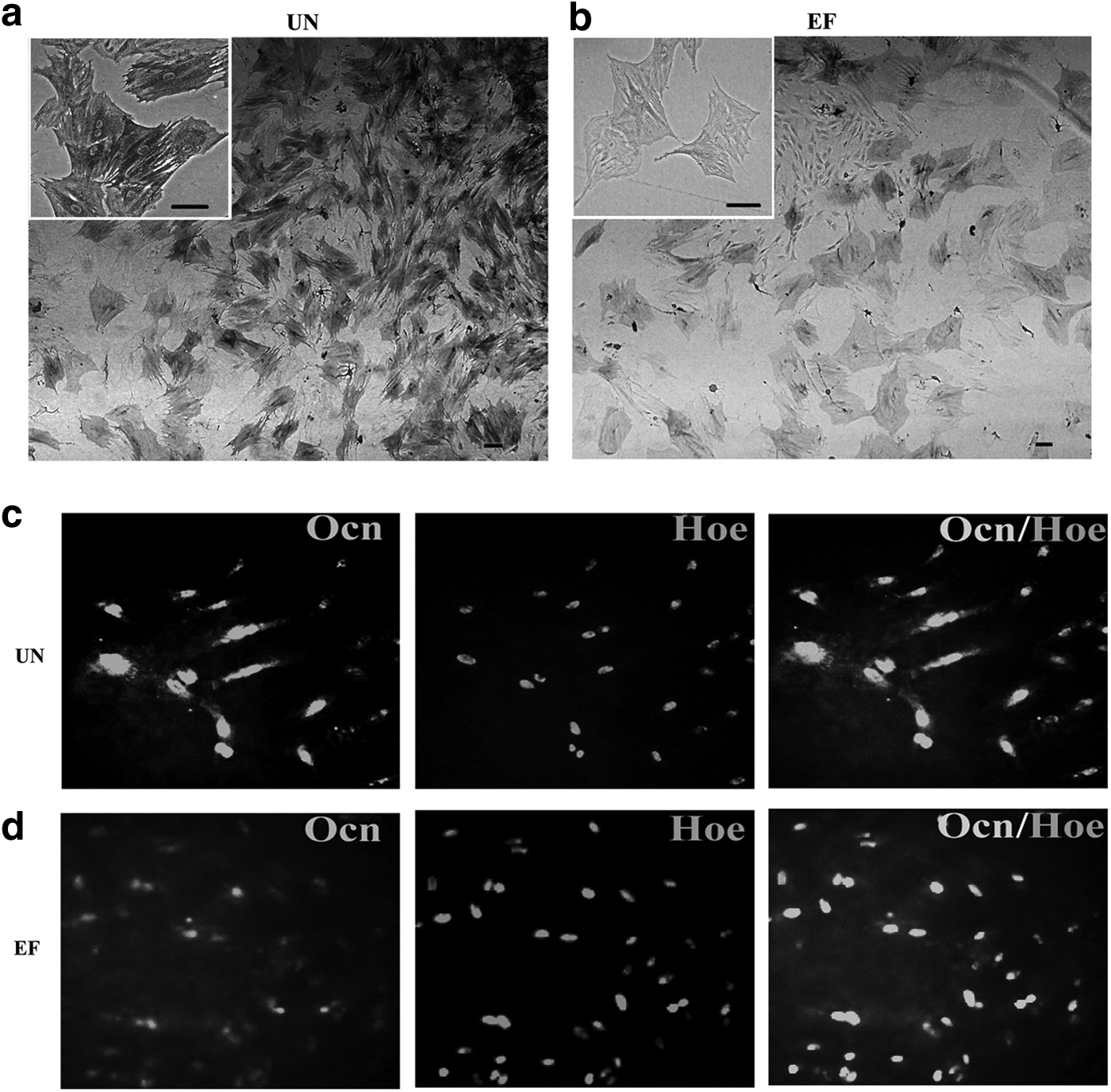
EGF and bFGF reduce ASC osteogenic differentiation. (
EGF and bFGF enhance the neural differentiation potential of ASCs
Next, the ability of ASCs cultured in UN or EF medium to differentiate into the neural lineage after the addition of RA was investigated. Morphologically, ASCs cultured in medium containing RA retracted continuously to form a sphere-shaped group of cells (Fig. 4a). Subsequently, the cells migrated from the sphere and formed simple bipolar-shaped cells. ASCs cultured in UN medium containing RA for 15 days expressed Nestin, an early neuronal marker, but did not express detectable levels of Gfap (Fig. 4b, d, f). Conversely, ASCs cultured in EF medium containing RA displayed retractile cell bodies with highly branched, complex multipolar structures and expressed Gfap and low levels of Nestin (Fig. 4c, e, g). Immunofluorescent staining confirmed that these markers were continuously expressed for at least 2 weeks, which indicated that ASCs cultured in EF neural differentiation medium terminally differentiated into neural lineage cells. In contrast, the mature neural proteins Gfap and Map2 were barely detectable in the cells cultured in UN neural differentiation medium. Nevertheless, the ASCs derived from EF neural differentiation medium expressed Gfap and Map2, but not Nestin. Quantification revealed that expression of Nestin decreased 9-fold, whereas Gfap and Map2 were upregulated 21.9-fold and 18.9-fold, respectively, in ASCs derived from EF neural differentiation medium, compared to UN neural differentiation medium (Fig. 4i).
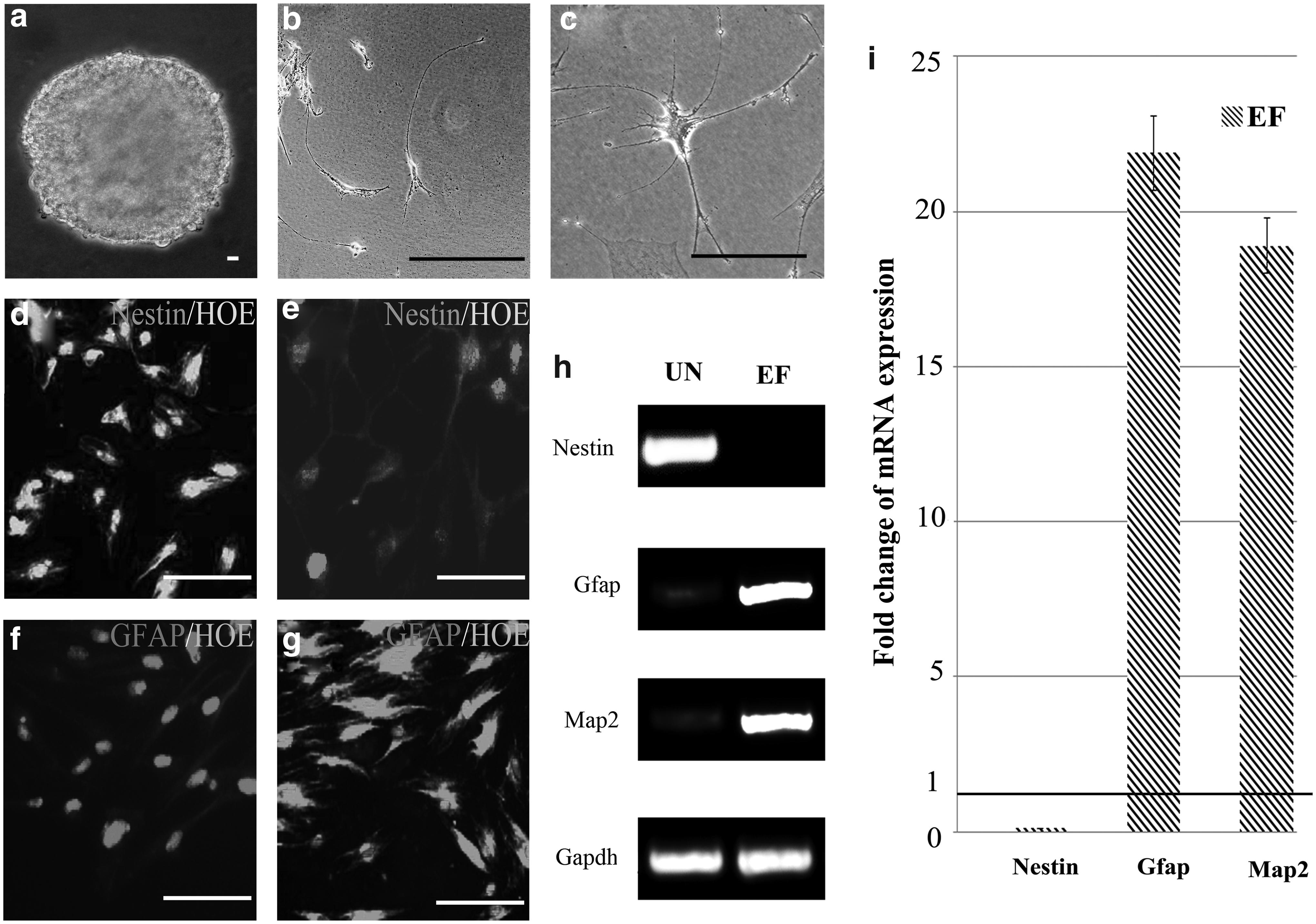
EGF and bFGF promote ASC neural differentiation. (
These data suggested that ASCs cultured in neural differentiation medium containing EGF and bFGF successfully transdifferentiated into mature neural cells, whereas the cells induced in UN neural differentiation medium only generated neural progenitors. These results show that ASCs cultured in EGF and bFGF will differentiate into neural cells rather than osteocyte lineage cells when treated with RA.
Discussion
Extracellular growth factors are involved in a number of biological signal–regulated kinase signaling and processes, which may affect cell adhesion, migration, proliferation, and differentiation (Discher et al., 2009; Lee et al., 2009). EGF and bFGF participate in a complex network of cellular processes, and are commonly added into stem cell cultures to enhance their proliferation capacity, with the expectation of maintaining the stem cells' ability for multipotent differentiation. Kakudo et al. and Kalyani et al. reported that high concentrations of EGF or bFGF may act as either positive or negative regulators of stem cell gene expression, as well as adipocyte, osteogenic, and chondrogenic differentiation (Kakudo et al., 2007; Kalyani et al., 1999). Meanwhile, other research indicated that incubation with low concentrations of EGF or bFGF significantly enhanced cell proliferation and migration, but did not affect the undifferentiated state of ESCs (Park et al., 2011; Vassaux et al., 1994). On the basis of these studies, the effects of low concentrations of a combination of EGF and bFGF on the proliferation and differentiation of ASCs during stem cell expansion in vitro were investigated.
ASC morphology, growth curves, and cell cycle analysis indicated that the addition of EGF and bFGF significantly increased the proliferative capacity of ASCs, and that these potent mitogenic activity factors were able to produce a greater quantity of ASCs (Hebert et al., 2009; Mydlo et al., 1998; Neubauer et al., 2004; Vassaux et al., 1994; Zuk 2010).
Oct4 and Sox2, in concert with other factors (Klf4, Nanog, Lin28a), are involved in the maintenance of stem cell self-renewal and pluripotency (Chen et al., 2008; Driessens et al., 2011; Thomson et al., 2011; Wang et al., 2012b). Immunofluorescent staining and PCR revealed that EGF and bFGF enhanced Sox2 expression and downregulated Oct4 expression, and that EGF and bFGF could affect the expression of stem cell–related genes in ASCs. There is ample evidence to demonstrate that downregulation of Oct4 and upregulation of Sox2 can affect the pluripotency of stem cells, stimulate differentiation to the ectoderm lineage, and improve induction efficiency (Thomson et al., 2011; Wang et al., 2012b).
The results in this study revealed that EGF and bFGF may induce ASCs to transinduce into an ectodermal lineage rather than a mesodermal lineage. Osteogenic differentiation assays confirmed that ASCs cultured with EGF and bFGF had weaker AP activity and lower levels of Ocn, Cbfa, and Col compared to the cells treated without EGF and bFGF. Moreover, the neurogenic differentiation assay proved that ASCs treated with EGF and bFGF preferred to transdifferentiate into mature neurons, even in the presence of low concentrations of EGF and bFGF. Expression of Gfap in ASCs induced in the presence of EGF and bFGF indicated that the cells had partially differentiated into glial cells.
This study demonstrates that EGF and bFGF play an important role in the proliferation and stem cell plasticity of ASCs. Low concentrations of EGF (5 ng/mL) and bFGF (10 ng/mL) not only enhanced the proliferation of ASCs, but also affected their differentiation. EGF and bFGF, even at low concentrations, may limit the differentiative ability of stem cells during stem cell expansion in vitro; therefore, the supplementation of stem cell cultures with EGF and bFGF should be carefully considered during in vitro expansion for clinical applications.
Footnotes
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (973 Program) (no. 2013CB932902), and the National Natural Science Foundation of China (NSFC) (no. 61071047)
Author Disclosure Statement
The authors declare that there is no conflict of interest.