
Abstract
Abstract
Previously, we successfully generated fully grown, cloned medaka (the Japanese rice fish, Oryzias latipes) using donor nuclei from primary culture cells of adult caudal fin tissue and nonenucleated recipient eggs that were heat shock-treated to induce diploidization of the nuclei. However, the mechanism of clone formation using this method is unknown, and the rate of adult clone formation is not high enough for studies in basic and applied sciences. To gain insight into the mechanism and increase the success rate of this method of clone formation, we tested two distinct nuclear transfer protocols. In one protocol, the timing of transfer of donor nuclei was changed, and in the other, the size of the donor cells was changed; each protocol was based on our original methodology. Ultimately, we obtained an unexpectedly high rate of adult clone formation using the protocol that differed with respect to the timing of donor nuclei transfer. Specifically, 17% of the transplants that developed to the blastula stage ultimately developed into adult clones. The success rate with this method was 13 times higher than that obtained using the original method. Analyses focusing on the reasons for this high success rate of clone formation will help to elucidate the mechanism of clone formation that occurs with this method.
Introduction
The study of nuclear transfer in fish was initiated by Tung and his colleagues in the 1960s, and production of nuclear transplants (NTs) with donor nuclei from adult cells remained unsuccessful for more than 40 years (Di Berardino, 1997). In the late 1990s, we began studying nuclear transfer in fish using medaka (the Japanese rice fish, Oryzias latipes), a small freshwater fish that is widely used as a model organism in the life sciences (Kinoshita et al., 2009; Takeda et al., 2011), to understand and overcome the obstacles to nuclear transfer of donor nuclei from adult fish.
Initially, we tried to transfer donor nuclei from embryonic cells to recipient nonenucleated unfertilized eggs; we obtained triploid transplants with donor genetic markers that we believed were formed by the fusion of donor diploid and recipient haploid nuclei (Niwa et al., 1999; Niwa et al., 2000). This method of nuclear transfer using nonenucleated recipient eggs was applied to studies of production of diploid animals by fusion of nuclei of donor haploid cells, such as haploid embryonic stem cells (ESCs) (Yi et al., 2009) or sperm (Liu et al., 2011), and recipient haploid nuclei of nonenucleated unfertilized eggs in medaka. In our studies of nuclear transfer with donor embryonic cells, we successfully produced diploid clones by transferring donor nuclei to enucleated unfertilized eggs in 2001 (Wakamatsu et al., 2001). In 2007, we successfully cloned fish for the first time using adult somatic cell nuclei as donors with this fish model (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008). In this method, donor nuclei from cells grown in primary cultures derived from adult caudal fin tissue were transferred into recipient eggs that were nonenucleated and that had been subjected to heat shock to induce diploidization of recipient egg nuclei. Approximately 1–3% of the reconstructed embryos that developed to the blastula stage successfully developed into diploid and fertile adult fish; detailed analysis of these fish revealed only donor genetic markers and no recipient markers. As a result, these fish were considered to possess only donor nuclei and, therefore, to be donor clones. However, mechanisms of clone formation using this method and of reprogramming of donor nuclei from adult tissue cells by this method remained unknown. Moreover, the success rate of adult clone formation with this method is not high enough to apply this method reliably to studies in the basic or applied fields of sciences.
Here, we experimented with modifications to our previously published method for nuclear transfer to gain insight into the mechanism of clone formation and to increase the success rate of clone formation; specifically, we transferred donor nuclei to recipient eggs that were prepared via distinct but related processes to those used in our previous method, and we explored the effect of donor cells size. Ultimately, we developed a modified protocol with an unexpectedly high success rate of adult fish clone production; this rate was 13 times higher than that with the original protocol. This new protocol may provide important clues for analysis of the mechanism of clone formation. Our findings may shed light, not only on fish cloning, but also on cloning of other animals, including mammals, and on mechanisms of cell reprogramming.
Materials and Methods
Breeding of experimental animals
Fish were maintained and bred as described previously (Niwa et al., 1999). Developmental stages were determined in accordance with Iwamatsu's descriptions (Iwamatsu, 2004).
Medaka strains
Strains used for the donor nuclei and the recipient eggs
Two strains of transgenic medaka, d-rR-Tg(OlMA1-DsRed2) (Fig. 1A) and d-rR-Tg(OlMA1-GFP) (Fig. 1B) (Kinoshita, 2004), were used as the donor strain and the recipient strain, respectively. The d-rR-Tg(OlMA1-DsRed2) fish carried DsRed2, a gene encoding a red fluorescent protein (RFP), driven by the medaka α-actin gene promoter. In these fish, expression of DsRed2 was initially weak in anterior somites at the 30-somite stage; however, intensity of RFP fluorescence increased after this stage. Red fluorescence was observed throughout subsequent embryonic development and throughout all adult stages. The other transgenic fish, d-rR-Tg(OlMA1-GFP) carried the green fluorescent protein (GFP) gene driven by the medaka α-actin gene promoter. In this strain, GFP expression became evident at the 12-somite stage, and GFP intensity increased after this stage. The expression pattern of GFP in the recipient strain was the same as that of DsRed2 in the donor strain.
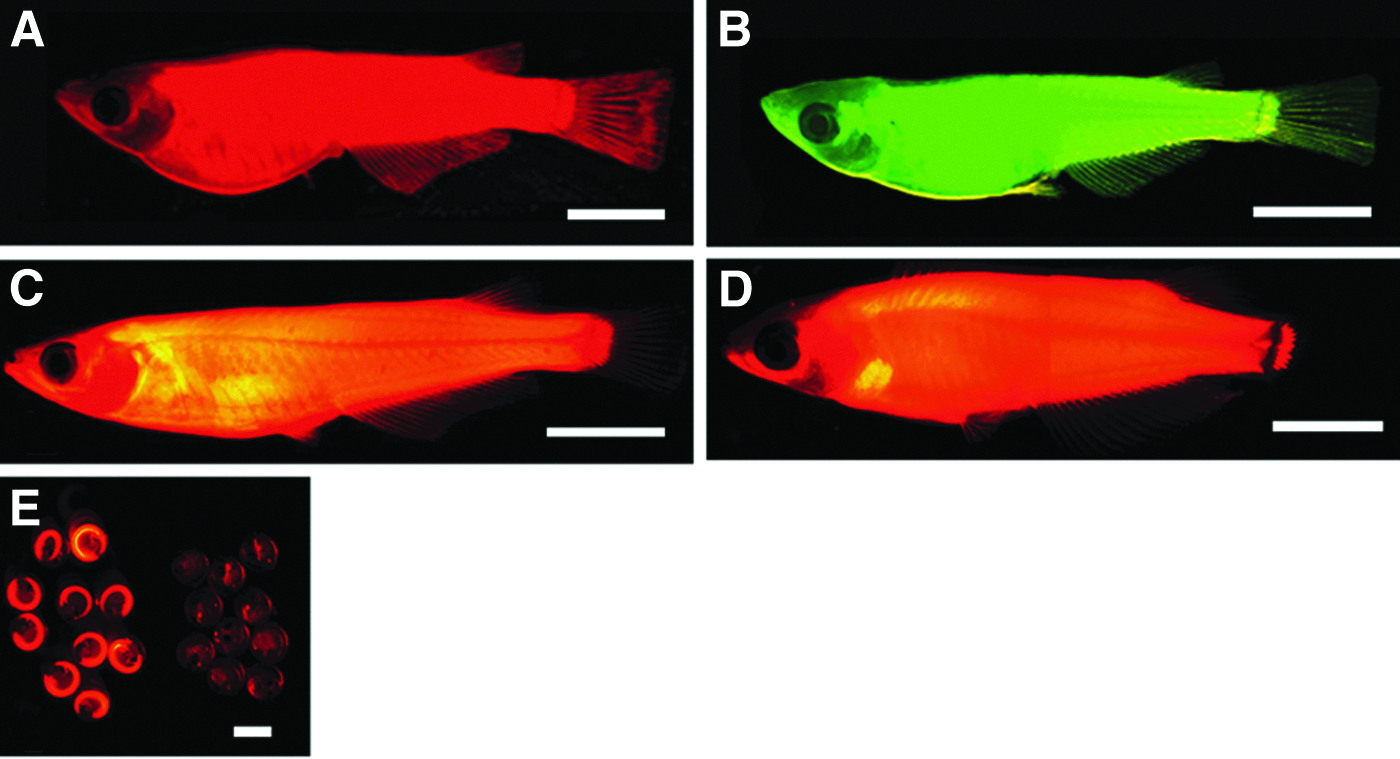
Images of fluorescence from the medaka strains used for nuclear transfer experiments and from a nuclear transplant and its progeny. (
The donor and recipient transgenic strains were each derived from d-rR (Yamamoto, 1953), a strain that lacks melanized melanophores in the skin because of the b/b genotype and shows sexual dimorphism in the phenotype of xanthophores in the skin because of sexually dimorphic genotypes of sex-linked r locus. The xanthophores are colorless in XrXr females, but they have wild-type pigment in XrYR males.
The strain used as the partner strain for genetic crosses
d-rR fish were used as mating partners for the NTs and their F1 progeny to assess the fertility of each NT and also to assess transmission of donor and recipient genetic markers to F1 and B1 progeny in genetic crosses (Fig. 2).

Genetic cross of a nuclear transplant and its F1 progeny with partner fish to assess the fertility of the nuclear transplant and also to assess transmission of the donor genetic marker to F1 and B1 progeny. The red fish is a carrier of the donor marker. NT, nuclear transplant; d-rR, partner fish strain. The B1 generation is presented as embryos.
Preparation of donor cells
The four distinct protocols used for nuclear transfer experiments are diagrammed in Figure 3. Primary cultures derived from the caudal fin tissues of a single adult donor fish were prepared as described previously (Bubenshchikova et al., 2008), and cells from these primary cultures were used as donor cells for a single nuclear transfer experiment. Our previous findings (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008) demonstrated that donor cells from either sex produce clones with the same sex as that of the donor. Although donor cells from females were used in a few experiments in this study, we usually used donor cells from males to demonstrate more clearly that the resultant clones originated from donor nuclei. When female cells were used as donors, the origin of the resultant NTs, which were females, was less clear because those female NTs could have originated under the influence of diploidized recipient nuclei contained in tissues of NTs. This is because the mechanism of removal of the diploidized recipient nuclei from the resultant transplants has not yet been resolved.
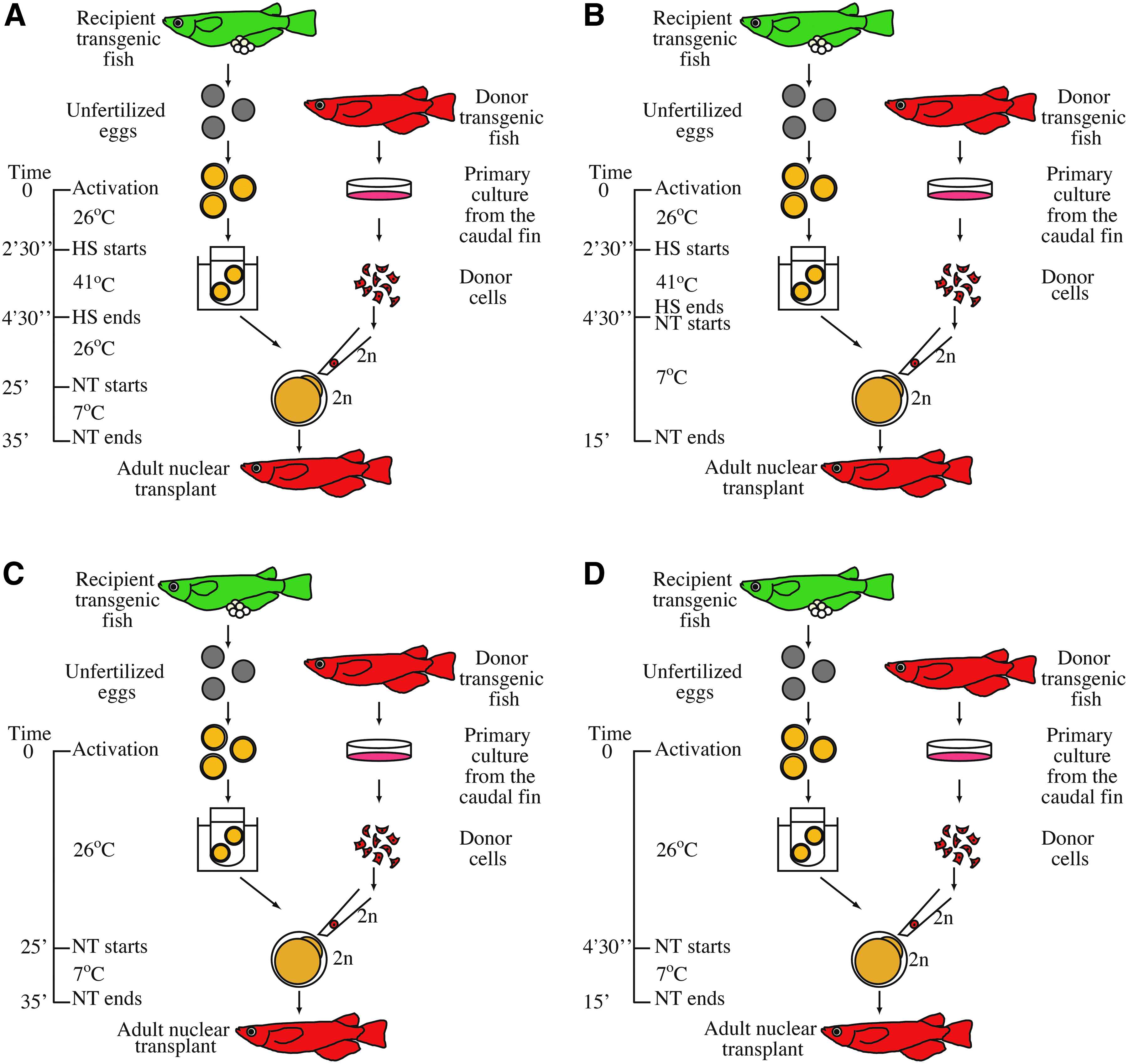
Diagrams showing protocols used in the nuclear transfer experiments. (
Cells of various and quite different sizes were observed in primary cultures from caudal fin tissues. The diameters of primary culture cells individuated by trypsinization ranged from 10 μm to 19 μm, and most of these cells had a diameter of approximately 12 μm. Of all the suspended cells, only large-sized (L) cells, those with diameters of 17–18 μm, were used as donor cells in our previous studies on adult somatic cell nuclear transfer. Here, we used L cells as donor cells in the Experiment 1 (Exp. 1) series—Exp. 1-1 and Exp. 1-2—and smaller (S) cells, those with diameters of 12–15 μm, in Exp. 2.
Preparation of recipient eggs
In the Exp. 1 series and in Exp. 2, recipient eggs were prepared via a method described previously (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008) with some modifications. Briefly, three to four unfertilized eggs collected from females of the recipient strain were activated with an electric pulse and incubated at 26°C for 2 min 30 sec in a balanced salt solution (BSS) designed for medaka (Iwamatsu, 1983). A previously described method was used to determine this incubation time for this particular recipient strain (Bubenshchikova et al., 2007). Specifically, we aimed to optimize conditions for diploidization of recipient nuclei. The eggs were then subjected to heat shock at 41°C for 2 min to induce diploidization of egg nuclei (Naruse et al., 1985). In Exp. 1-1 (Fig. 3A), the eggs thus prepared were incubated in BSS at 26°C for 20 min to allow the eggs to recover from heat shock. The eggs were then transferred into BSS at 7°C on an agar plate for nuclear transfer within 10 min. This process was repeated several times for a single experiment. This protocol was basically the same protocol used in our previous studies (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008).
In Exp. 1-2 and Exp. 2 (Fig. 3B), we eliminated the incubation step at 26°C for 20 min intended for recovery from heat shock; specifically, the eggs were transferred into BSS at 7°C on the agar plate for nuclear transfer directly after the heat shock treatment, and used for nuclear transfer within 10 min. This study was the first time we used this protocol to prepare eggs for nuclear transfer.
In addition, because this was the first time we used these particular strains as the donor and recipient strains, we examined the results under three control experimental conditions (i.e., those in Exp. 1-1C, Exp. 1-2C, and Exp. 2C) to assess the impact of the heat shock treatment that was used to induce diploidization of the egg nuclei on the success of nuclear transfer with these fish strains. The experimental conditions in Exp. 1-1C, Exp. 1-2C, and Exp. 2C (Fig. 3C, D) were based on those in Exp. 1-1, Exp. 1-2, and Exp. 2, respectively. However, in these control experiments, the recipient eggs were not subjected to heat shock treatment; instead, they were kept at 26°C, rather than 41°C, for 2 min
Nuclear transfer and observation and imaging of GFP and RFP signals
Nuclear transfer and observation and imaging of GFP and RFP signals were performed as described previously (Bubenshchikova et al., 2008).
Ploidy estimations
Ploidy estimates were performed as described previously (Bubenshchikova et al., 2008). The orange-red variety of medaka was used as the diploid control.
PCR analysis
The Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, St. Louis, MO, USA) was used according to the manufacturer's instructions to extract genomic DNA from the caudal fin tissues of adult fish or from whole embryo bodies. Primers specific for Tg(OlMA1-GFP) (forward, 5′-ATGGTGAGCAAGGGCGAGGAGCTG-3′; reverse, 5′-CTTGTACAGCTCGTCCATGCCGAG-3′) were used to amplify a 717-bp GFP gene fragment as described previously (Bubenshchikova et al., 2005). Similarly, primers specific for Tg(OlMA1-DsRed2) (forward, 5′-ATGGCCTCCTCCGAGAACGTC-3′; reverse, 5′-GTCCAGCTTGGCGTCCACGTA-3′) were used to amplify a 600-bp DsRed2 gene fragment. To confirm the success of DNA extraction and of PCR amplification, the endogenous elongation factor 1α-A gene (EF-1α-A) was also amplified using the following primers: forward, 5′-CAGGACGTCTACAAAATCGG-3′; reverse, 5′-AGCTCGTTGAACTTGCAGGCG-3′; the expected size of this PCR product was 519 bp. Using the present PCR protocol, the smallest detectable concentration of DNA was 0.04% (Bubenshchikova et al., 2008).
This study was performed according to the Guide for the Care and Use of Laboratory Animals by the Committee on Animal Experiment of the Bioscience and Biotechnology Center, Nagoya University.
Results
Findings from Exp. 1-2 and Exp. 2, each of which had unique and new experimental conditions, were compared with those from Exp. 1-1 because the protocol used in Exp. 1-1 was nearly identical to the protocol used in our previous studies (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008). Development of NTs generated in each of these experiments is summarized in Table 1.
The number in parentheses represents the percentage with respect to the total number of embryos that developed to the blastula stage.
GFP, green fluorescent protein.
Nuclear transfer using L cells as donors
L cells with diameters of 17–18 μm were used as donors in Exp. 1-1 (Fig. 3A) and Exp. 1-2 (Fig. 3B). In Exp. 1-1, which included a heat shock and a 20-min recovery period for recipient eggs, 343 nuclear transfers were attempted. From these 343 attempts, 156 transplants developed to the blastula stage; 46 (29.5% of 156) of these blastula embryos developed to the 30-somite stage, and only 5 (3.2% of 156) of the 46 embryos hatched. Of the five hatchlings, two exhibited only fluorescence from DsRed2, the donor genetic marker, and no fluorescence from GFP, the recipient genetic marker. Fluorescence from both DsRed2 and GFP was observed in another two hatchlings; the last hatchling showed only fluorescence from GFP and none from DsRed2. Of the five hatchlings, three animals—2 (1.3% of 156) with only DsRed2 fluorescence and one with only GFP fluorescence—grew normally and reached adulthood within 2 months after hatching. The expression pattern of DsRed2 fluorescence in the transplants that exhibited DsRed2 fluorescence alone was similar to that of donor fish and that of GFP fluorescence in the transplant that exhibited GFP fluorescence alone was similar to that of recipient fish throughout the embryonic and adult stages.
In Exp. 1-2, from which the 20-min recovery period from heat shock was omitted, 300 nuclear transfers were attempted; of these, 65 reached the blastula stage. The percentage (21.7%; 65/300×100) of embryos that developed to the blastula stage among all reconstructed embryos in this experiment was lower than a half of that (45.5%; 156/343×100) in Exp. 1-1. We believe that this low rate of successful development of reconstructed embryos to the blastula stage in Exp. 1-2 was due to omission of the recovery process, specifically omission of the 20-min period at 26°C to allow recovery from heat shock. However, the omission of this process had no apparent negative effects on the development of the blastula embryos to later stages; 24 (36.9% of 65) of these blastula embryos developed to the 30-somite stage, and 13 (20.0% of 65) of the 24 embryos developed further and hatched. Of these 13 hatchlings, 12 exhibited only fluorescence from DsRed2 with the expression pattern similar to that of donor fish and none from GFP. The one other hatchling exhibited chimeric expression of both DsRed2 and GFP. Of the 12 hatchlings exhibiting only DsRed2 fluorescence, 11 (16.9% of 65) grew normally to adulthood (Fig. 1C); in contrast, the 12th one died within 2 weeks after hatching. The chimeric hatchling that exhibited both DsRed2 and GFP fluorescence also died within a week after hatching. The success rate of nuclear transfer, specifically, the percentage of the number of adult NTs that expressed only the donor marker among all embryos that reached the blastula stage, in this experiment was 13.2 times higher than that in Exp. 1-1. When the success rate was expressed as the percentage of adult NTs that expressed only the donor marker among all reconstructed embryos, the success rate in Exp. 1-2 was 6.3 times higher than that in Exp. 1-1.
Among the embryos that developed to the 30-somite stage in Exp. 1-2, 66.7% (16/24×100) exhibited only DsRed2 fluorescence and 16.7% (4/24×100) each exhibited chimeric expression of DsRed2 and GFP or only GFP expression; however, 8.7% (4/46×100) of the 30-somite stage embryos from Exp. 1-1 exhibited only DsRed2 fluorescence, but 63.0% (29/46×100) were chimeric, and 28.3% (13/46×100) exhibited GFP fluorescence alone. Thus, the percentage of embryos at the 30-somite stage with only DsRed2 fluorescence in Exp. 1-2 was 7.7 times higher than that in Exp. 1-1.
From the first evidence of donor or recipient marker fluorescence (DsRed2 or GFP, respectively) in each NT embryo, the expression pattern of fluorescence did not change throughout the life of the animal; that is, a NT embryo with DsRes2 fluorescence alone grew to be an adult NT with only DsRes2 fluorescence, and an embryo with GFP fluorescence alone or with chimeric expression of DsRed2 and GFP grew to be an adult with GFP fluorescence alone or an adult with chimeric expression, respectively. This stability in gene expression was the same in each trial, regardless of whether it was an experimental or control trial.
Development of reconstructed embryos in the control experiments, Exp. 1-1C (Fig. 3C) and Exp. 1-2C (Fig. 3D), is summarized in Table 2; these two control experiments were designed to assess the impact of the heat shock treatment on the success of nuclear transfer, that is, in Exp. 1-1C and Exp. 1-2C, the heat shock treatment was omitted, but otherwise the protocols used were identical to those used for Exp. 1-1 and Exp. 1-2, respectively. The percentage (43.5%; 154/354×100) of embryos that developed to the blastula stage among all of the reconstructed embryos in Exp. 1-2C was higher than that in Exp. 1-2 (21.7%). We believe that this higher percentage of embryos developing to the blastula stage resulted from the omission of the heat shock treatment in the control experiment. The percentage of embryos that developed to the blastula stage among all of the reconstructed embryos was 42.2% (149/353×100) in Exp. 1-1C and 43.5% in Exp. 1-2C); these percentages are remarkably similar to each other. This result means that the difference in the timing of transfer of the donor nuclei to the recipient eggs after the electric pulse activation of recipient eggs in these two experiments did not have any apparent effects on the development of the reconstructed embryos to the blastula stage. These percentages in Exp. 1-1C and Exp. 1-2C were also similar to that in Exp. 1-1 (45.5%).
The numbers in parentheses represent the percentage with respect to the total number of embryos that developed to the blastula stage.
GFP, green fluorescent protein.
In Exp. 1-1C, the percentage (28.2%; 42/149×100) of embryos that developed to the 30-somite stage among the embryos that developed to the blastula stage was similar to that in Exp. 1-1. However, all of the embryos exhibited chimeric DsRed2 and GFP fluorescence or only GFP fluorescence, and no embryo exhibited DsRed2 fluorescence alone. As the result, no hatchling or adult with only DsRed2 fluorescence was recovered. A small number of hatchlings with chimeric expression of DsRed2 and GFP or expression of only GFP were recovered; however, all of these animals died within 2 weeks after hatching. Similar results were obtained in Exp. 1-2C, though no hatchlings were recovered. Therefore, no adults were recovered in either of the control experiments 1-1C or 1-2C.
Nuclear transfer using S cells as donors
The protocol used for Exp. 2 (Fig. 3B) was the same as that used for Exp. 1-2 in that neither of the protocols included a 20-min recovery period for heat shock; however, in Exp. 2, only S cells with diameters of 12–15 μm were used as donors, and 302 nuclear transfers were attempted. From these 302 attempts, 70 (23.2%; 70/302×100) transplants developed to the blastula stage. This percentage of development to the blastula stage among all reconstructed embryos was lower than that in Exp. 1-1 and was similar to that in Exp. 1-2. We believe that this reduced survival to the blastula stage resulted from omission of the 20-min recovery period at 26°C. As in Exp. 1-2, the omission of this heat shock recovery period had no apparent negative effects on the development of blastula embryos to later stages; 30 (42.9% of 70) such blastulas developed to the 30-somite stage.
Among the 30 embryos that developed to the 30-somite stage, 3 (10% of 30) expressed only DsRed2, 17 (56.7% of 30) expressed DsRed2 and GFP in a chimeric pattern, and 10 (33.3% of 30) expressed only GFP. Of these 30 embryos, six (8.6% of 70) developed further and hatched; three of these six hatchlings expressed only DsRed2 with a similar pattern to that of donor fish, and the other three expressed DsRed2 and GFP in a chimeric pattern. Of the six hatchlings, two reached adulthood; one (1.4% of 70) exhibited only DsRed2 fluorescence (S. 1A), and the other one exhibited chimeric expression of DsRed2 and GFP (S. 1B). In this experiment, the percentage (1.4%) of adult NTs that expressed only the donor marker of all embryos that reached the blastula stage was similar to that (1.3%) in Exp. 1-1, but not to that (16.9%) in Exp. 1-2.
In the control experiment Exp. 2C (Fig. 3D), in which the heat shock treatment was omitted from the protocol of Exp. 2, the percentage (33.5%; 85/254×100) of embryos that developed to the blastula stage among all of the reconstructed embryos was higher than that (23.2%; 70/302×100) in Exp. 2 (Table 2). The omission of the heat shock treatment in the Exp. 2C was considered to be the reason for the higher percentage of embryos that developed to the blastula stage.
None of the 25 embryos that developed to the 30-somite stage expressed DsRed2 alone, although all of them expressed both DsRed2 and GFP in a chimeric pattern or GFP alone. One of the embryos that exhibited only GFP fluorescence hatched, but it died within a week after hatching. As a result, no adult fish was obtained in this control experiment.
Characterization of NTs
Characteristics of each adult NT obtained in the present study were analyzed and the findings are summarized in Table 3. For each of the 14 adult NTs that expressed only DsRed2 and were recovered in Exp. 1 or Exp. 2, i.e., 2 (1-1NT2, 1-1NT3) in Exp. 1-1, 11 (1-2NT1 through 1-2NT11) in Exp. 1-2, and 1 (2NT1) in Exp. 2, the DsRed2 gene was readily detected via PCR analysis, but the GFP gene was undetectable (Fig. 4A, S. 2). Conversely, the GFP gene was detected in 1-1NT1, an adult that exhibited only GFP fluorescence, but the DsRed2 gene was not (S. 2). In the chimeric NT, 2NT2, both DsRed2 and GFP genes were detected in the PCR analysis (data not shown).
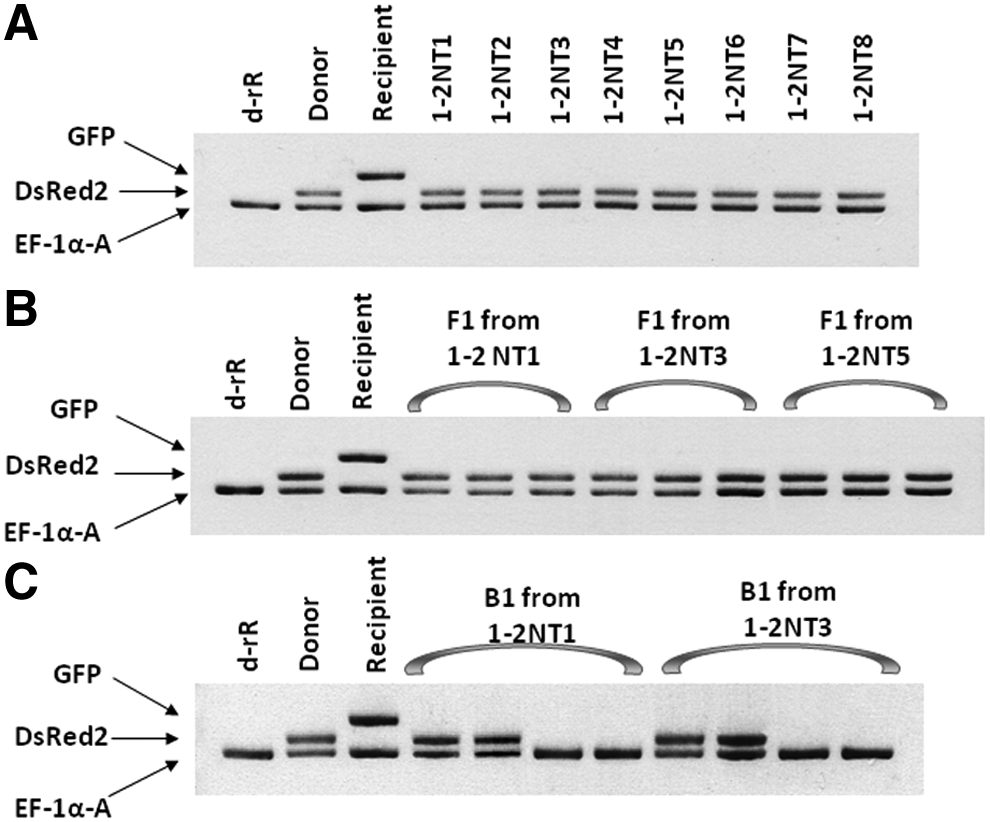
PCR analyses for detection of DsRed2 and GFP in nuclear transplants in Exp. 1-2 and their F1 and B1 progeny. The DsRed2 gene was evident in all nuclear transplants and F1 progeny and in each of the B1 progeny that exhibited DsRed2 fluorescence, but the GFP gene was not evident in any of the nuclear transplants or their F1 or B1 progeny. (
Weakly positive.
NT, nuclear transplant; GFP, green fluorescent protein; F, female; M, male; ND, not determined.
Ploidy analysis revealed that each of the 13 adult NTs examined was diploid (S. 3). The sex of NTs was determined by the secondary sexual characteristics and their reproductive behavior. Each NT, except that of 1-1NT1 and of 2NT2, was the same sex as that of the respective donor, that is, male NTs were obtained from donor male nuclei and females from donor female nuclei. 1-1NT1 and 2NT2 were both female, but the respective donor nuclei each originated from a male. These individuals, 1-1NT1 and 2NT2, expressed GFP florescence alone or both DsRed2 and GFP florescence. This finding suggested that all or a part of each of their bodies was composed of female cells that originated from the diploidized recipient nuclei and that these female cells, but not the male cells originating from donor male nuclei, affected sex determination in these fish.
Each NT was mated to partner fish from the d-rR strain (Fig. 2). Only one NT, 1-1NT3, did not produce any fertilized eggs, and the chimeric NT (2NT2) was only weakly fertile. Additionally, two NTs, 1-2NT7 and 1-2NT11, died before they could be crossed with partners. Each of the other 12 NTs showed normal fertility.
Transmission of genetic markers from NTs to their progeny
To determine whether each fertile NT transmitted donor or recipient genetic markers to F1 and B1 progeny in genetic crosses to partner d-rR fish, each of the 12 NTs with normal fertility was used for a separate genetic cross (Fig. 2). The findings of these crosses are summarized in Table 4. In the genetic analysis of progeny from the 11 NTs that carried only the DsRed2 donor marker gene (1-1NT2, 1-2NT1 through 1-2NT6, 1-2NT8 through 1-2NT10, and 2NT1), between 212 and 894 F1 progeny were collected from each cross between a single NT individual and a partner d-rR fish. Each of these F1 progeny expressed only DsRed2 fluorescence in a pattern identical to that in the parental NT (Fig. 1D). On the basis of PCR analysis, only the DsRed2 gene, but not the GFP gene, was detected in each F1 individual from these 11 crosses (Fig. 4B). For each of the fertile 11 NTs, five, six, or seven pairwise “F1 crosses” were set up; in each “F1 cross,” and an individual F1 animal was mated to a d-rR partner fish. Between 766 and 1919 B1 progeny were collected from each of these “F1 crosses.” The B1 generation from each “F1 cross” included progeny with the donor marker gene and fluorescence from the gene product and progeny without the marker gene or the associated fluorescence in an apparent 1:1 ratio (Fig. 1E and Fig. 4C).
Numbers in parentheses represent the percentage with respect to the total number of collected embryos of F1 or B1 progeny.
NT, nuclear transplant; GFP, green fluorescent protein; F, female; M, male; ND, not determined.
All 311 F1 progeny from 1-1NT1 expressed only the recipient marker GFP fluorescence. Each of them carried the GFP gene, but not the DsRed2 gene (data not shown). Only half of the 703 B1 progeny from this NT exhibited GFP fluorescence; the other half did not. The GFP gene was detected in the progeny with the GFP fluorescence, but not in those without GFP fluorescence (data not shown). The chimeric NT, 2NT2, gave rise to only a small number of F1 progeny with green fluorescence alone. This fact suggested that germ cells that originated from the diploidized recipient nucleus were contained in the ovaries of this fish. We did not attempt any genetic crosses with the F1 progeny.
Thus, for each NT with normal fertility, the donor or recipient marker gene was transmitted to the respective F1 and B1 progeny in an apparently Mendelian fashion.
Discussion
Previously, we developed a method for nuclear transfer in medaka that used donor nuclei from cells from primary cultures of adult caudal fin tissue and recipient nonenucleated eggs that had been subjected to heat shock to induce diploidization of recipient egg nuclei. This method successfully produced nuclear transplants that were diploid and fertile and that showed donor genetic characteristics and no recipient genetic characteristics (Bubenshchikova et al., 2007; Bubenshchikova et al., 2008). On the basis of the characteristics of these transplants, they were considered to be clones that had originated from the donor nuclei, in spite of the use of nonenucleated eggs as recipients. The fact that donor clones were produced using this method indicated that the donor nuclei had been reprogrammed in the cytoplasm of reconstructed embryos and that the body of the transplants had formed from cells that had originated from the donor nuclei and not from cells that had originated from the recipient nuclei. Moreover, these findings indicated that the donor and recipient nuclei behaved independently of each other and did not fuse in the reconstructed embryos. However, the mechanism of clone formation in this method remains unknown. To gain insight into the mechanism of clone formation and to increase the success rate of clone formation, we tried two new protocols for nuclear transfer; each was a partially modified version of the original method.
The original protocol was used in Exp. 1-1 and resulted in a success rate for clone formation that was comparable with the rates observed in previous studies. In both experiments with modified experimental conditions (Exp. 1-2 and Exp. 2), diploid and fertile adult fish with only the donor genetic marker were recovered. These adult fish were healthy and transmitted the donor marker to their progeny in an apparently Mendelian fashion. Therefore, we conclude that these NT fish were clones of the donor fish. However, the rates of adult clone formation were very different between these two experiments. In Exp. 1-2, L cells were used as the donors and were transferred to recipient egg cytoplasm just after heat shock treatment and adult clones were obtained at a high rate: 17% of the embryos that developed to the blastula stage developed into adults. This success rate was 13 times higher than that with the original method (Exp. 1-1).
Interestingly, with the new protocol of Exp. 1-1, 66.7% of the embryos that developed to the 30-somite stage showed only the donor genetic marker and were considered to be clones. This percentage was 7.7 times higher than that in Exp. 1-1. The high percentage of clonal embryos at the 30-somite stage led to the high rate of adult clone formation. This result indicated the condition specific to Exp. 1-2, specifically the transfer of donor nuclei to the recipient eggs just after heat shock treatment, had an effect, and it preferentially promoted reprogramming of donor adult somatic cell nuclei to totipotency and development of clone embryos.
In Exp. 2, nuclei from S cells were transferred into recipient eggs that had been prepared via the same protocol used in Exp. 1-2. However, the success rate in this experiment was not as high as that in Exp. 1-2; instead, it was similar to that in the Exp. 1-1. Moreover, in Exp. 2, only 10% of the embryos that developed to the 30-somite stage expressed only the donor marker and were considered to be clones; the others expressed only the recipient marker or both the donor and recipient markers in a chimeric pattern. The percentage of clones at this developmental stage was also comparable to that in Exp. 1-1. This result shows that L cells were more suitable than S cells as donors for the development of clones via nuclear transfer. Thus, the results from Exp. 1-1, Exp.1-2, and Exp. 2 indicated that the combination of specific conditions in donor nuclei and recipient eggs in Exp. 1-2 promoted the preferential development of donor-derived clones.
No transplant embryo with the donor marker alone was recovered in Exp. 1-1C, Exp. 1-2C, or Exp. 2C; therefore, no hatchlings or adult fish with only the donor marker were recovered. These three control experiments were conducted to assess the effectiveness of the heat shock treatment. These findings indicated that heat shock treatment of the recipient eggs was essential for development of clone embryos and that the heat shock induced an activity in the recipient eggs that led to reprogramming of the donor adult somatic cell nuclei.
The results in Exp. 1-1 and Exp. 1-2 indicated that an activity in the recipient eggs that is necessary for reprogramming of donor nuclei was high just after the heat shock treatment of the recipient eggs and that this reprogramming activity declined over time. This reprogramming activity might come from some factors or conditions or a combination of factors and conditions in the recipient eggs that are optimal just after the heat shock treatment. Heat shock treatment induced diploidization in the recipient nuclei. Some aspects of the diploidized eggs might be similar to those of zygotes. Zygotes are known to play a role in reprogramming of sperm genomes (Rideout III et al., 2001). Egli and Eggan (2010) showed that nuclear factors in recipient zygotes can reprogram donor nuclei in mice nuclear transfer.
The cell cycle phase of both the donor and the recipient may be critical to successful animal cloning (Campbell et al., 1996; Wakayama, 2000). Wilmut et al. (1997) produced the first cloned animal, Dolly the sheep, using a donor adult somatic cell nucleus at the G0 phase and a recipient oocyte in metaphase II of meiosis. A somatic donor nucleus in interphase must undergo various mitotic processes following transfer to the recipient egg, and before the first cleavage for successful development of reconstructed embryos can occur. These processes take time, and cell cycle lengths during early cleavage stages in recipient eggs are generally too short for the necessary processes to occur in donor nuclei. Egli et al. (2007) averted this problem and generated mouse clones by transferring donor nuclei from mitotic cells into recipient zygotes that were in interphase; in the mitotic donors, DNA replication and metaphase chromosome formation had been completed at the time of transfer. The donor and recipient became synchronized with respect to cell cycle phase at the first cleavage.
In fish and other nonmammalian vertebrates, the cell cycle length during the early divisions of recipient eggs is often much shorter than that of donor somatic cells; therefore, cell cycle compatibility between donor and recipient cells is a significant concern in efforts to clone nonmammalian vertebrates
In Exp. 1-2, donor nuclei were transferred to the recipient eggs immediately after the heat shock treatment, whereas in Exp. 1-1, they were transferred to the recipients after a 20-min recovery from heat shock. At 18°C, first cleavage of medaka zygotes occurs within 90 min after adding sperm to eggs (Yamamoto, 1975), and metaphase chromosome formation occurs with the same timing in eggs activated via electric pulses as in sperm-activated eggs (E. Kaftanovskaya and Y. Wakamatsu, unpublished data). Less time elapses between heat shock treatment of recipient eggs and nuclear transfer in Exp. 1-2 than in Exp. 1-1; therefore, the transferred nucleus has more time in Exp. 1-2 than in Exp. 1-1 to progress through the various mitotic processes before a reconstructed embryo begins to cleave. The additional time between nuclear transfer and initiation of cleavage divisions in Exp. 1-2 may be important to normal development of the reconstructed embryos.
Mature, unfertilized medaka eggs are arrested in metaphase II of meiosis. These eggs can be released from this arrest via electric pulse activation; the activated eggs then progress continuously through the meiotic cell cycle and can be used as recipient eggs in nuclear transfer experiments as in the present study. In mammalian species, oocytes at metaphase II have been used as recipients of transferred donor somatic cell nuclei because the metaphase cytoplasm of these unfertilized recipient eggs is thought to reprogram somatic cell nuclei and confer totipotency to the donor nuclei (Gurdon and Wilmut, 2011; Tani et al., 2003). Mitotic zygotes can also reprogram transferred nuclei (Egli et al., 2007; Egli and Eggan, 2010). To better understand the high rate of clone formation in Exp. 1-2, it might be important to know where the recipient eggs are in the cell cycle at the time of nuclear transfer and whether the cytoplasm of these eggs has reprogramming activity
Recipient eggs were kept at 7°C after heat shock in Exp. 1-2, but they were kept at 26°C while the eggs recovered from the heat shock treatment in Exp. 1-1. The effects of low temperature after heat shock on the diploidization of recipient eggs are unknown. For example, cold shock, like heat shock or pressure, can inhibit release of the second polar body in fish eggs and thereby induce diploidization in haploid eggs (Gervai et al., 1980; Swarup, 1956; Swarup, 1959). Whether keeping eggs at 7°C after heat shock treatment in Exp. 1-2 promoted diploidization of nuclei in recipient eggs is an interesting possibility; previously, we demonstrated that the rate of successful adult clone formation was higher in the experiments that involved recipient eggs with a higher rate of diploidization via an optimally timed heat shock treatment than in experiments that involved eggs with a lower rate of diploidization via a nonoptimally timed heat shock step (Bubenshchikova et al., 2007).
We gained some insight into the mechanism of clone formation that occurs with the methods we have developed. Future analyses focusing on the reasons for the high success rate of clone formation achieved in the present study will help to elucidate the mechanism of clone formation that occurs with this method. A better understanding of this mechanism might lead to new insights into clone formation and into the reprogramming of adult somatic cells to pluripotency, not only in fish, but also in other vertebrates such as mammals.
Footnotes
Acknowledgments
We thank C. Inoue for providing technical assistance. This work was supported by the Program for the Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.