
Abstract
Abstract
The somatic cell nuclear transfer (SCNT) technique has been widely applied to clone pigs or to produce genetically modified pigs. Currently, this technique relies mainly on using terminally differentiated fibroblasts as donor cells. To improve cloning efficiency, only partially differentiated multipotent mesenchymal stem cells (MSCs), thought to be more easily reprogrammed to a pluripotent state, have been used as nuclear donors in pig SCNT. Although in vitro–cultured embryos cloned from porcine MSCs (MSCs-embryos) were shown to have higher preimplantation developmental ability than cloned embryos reconstructed from fibroblasts (Fs-embryos), the difference in in vivo full-term developmental rate between porcine MSCs-embryos and Fs-embryos has not been investigated so far. In this study, we demonstrated that blastocyst total cell number and full-term survival abilities of MSCs-embryos were significantly higher than those of Fs-embryos cloned from the same donor pig. The enhanced developmental potential of MSCs-embryos may be associated with their nuclear donors' DNA methylation profile, because we found that the methylation level of imprinting genes and repeat sequences differed between MSCs and fibroblasts. In addition, we showed that use of transgenic porcine MSCs generated from transgene plasmid transfection as donor cells for SCNT can produce live transgenic cloned pigs. These results strongly suggest that porcine bone marrow MSCs are a desirable donor cell type for production of cloned pigs and genetically modified cloned pigs via SCNT.
Introduction
Donor cell type is a major factor that determines the efficiency of SCNT. Current pig cloning is primarily using fibroblasts (Fs) as donor cells (Vajta et al., 2007). This terminally differentiated donor cell type is believed to frequently cause inappropriate nuclear reprogramming in SCNT embryos and thus results in a low birth rate of cloned animals (Cho et al., 2007). To increase the ability to generate cloned pigs, a less differentiated donor cell type, mesenchymal stem cells (MSCs), has been tested with SCNT in pigs. It was reported that in cultured porcine embryos cloned from MSCs (MSCs-embryos), the expression pattern of developmentally important genes involved in pluripotency, epigenetic modification, imprinting, and apoptosis was more similar with that of in vivo–derived embryos, as compared with embryos cloned from Fs (Fs-embryos) (Kumar et al., 2007). Moreover, several studies have reported that the in vitro preimplantation developmental potential of porcine MSCs-embryos was significantly higher than that of Fs-embryos (Colleoni et al., 2005; Faast et al., 2006; Jin et al., 2007; Kumar et al., 2007; Lee et al., 2010).
However, to our knowledge no study has been published so far aiming to compare the in vivo full-term developmental ability between MSCs-embryos and Fs-embryos. To fully evaluate the potential of MSCs in improvement of pig cloning efficiency, we used bone marrow MSCs and ear fibroblasts isolated from the same donor pig as donor cells to generate two different types of SCNT embryos and compared their in vitro preimplantation and in vivo full-term developmental rates. We also compared the DNA methylation status of repeat elements and genes involved in pluripotency and imprinting between porcine MSCs and fibroblasts. Furthermore, we investigated whether the porcine bone marrow MSCs can be used as donor cells to create transgenic pigs by the SCNT approach.
Materials and Methods
Ethics statements
This study was carried out in strict accordance with “The Instructive Notions with Respect to Caring for Laboratory Animals” issued by the Ministry of Science and Technology of China. The animal experimental protocol was approved by the Institutional Animal Care and Use Committee of South China Agricultural University. All efforts were made to minimize animal suffering.
Isolation of bone marrow MSCs and ear fibroblasts from the same donor pig
Three- to 5-day-old male piglets were used as donors for isolation of bone marrow MSCs and ear fibroblasts. Gelatinous bone marrow was extracted from the femur by a 16-gauge needle. The extracted bone marrow was layered on a Ficoll-paque gradient (density=1.077g/mL; Amersham Biosciences, Sweden) and centrifuged at 400×g for 22 min at room temperature. The mononuclear cells on the interface buffy layer were isolated and resuspended in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and cultured in 5% CO2 at 38°C. Nonadherent cells were gently removed 2 days after plating. Once they reached 80–90% confluence, the attached cells (passage 0) were dissociated with 0.25% (wt/vol) trypsin-EDTA solution and centrifuged at 300×g for 5 min. The cells were then passaged at a density of 1×105/cm2.
Ear fibroblasts were isolated as described by Deng et al. (2011). Isolated fibroblasts were cultured in DMEM supplemented with 10% (vol/vol) FBS at 39°C in a humidified atmosphere of 5% CO2 and 95% air.
Characterization of cell-surface markers of isolated MSCs
The passage-3 MSCs were trypsinized and fixed with 4% paraformaldehyde solution for 30 min. Nonspecific binding was prevented by incubating the cells with 3% goat serum and 0.3% Triton X-100 in Dulbecco's phosphate-buffered saline (D-PBS) for 30 min. Cells then were washed twice with D-PBS, incubated in 15 μg/mL of the primary anti-CD29, or anti-CD44, or anti-CD45, or anti-CD105 antibody or control isotype immunoglobulin G (IgG; BD Pharmingen, NJ, USA) for 30 min at room temperature. After washing with D-PBS, cells were incubated with a 1:500 dilution of Alexa Fluor 488 goat anti-mouse IgG for 20 min. Following washing with D-PBS, fluorescent cell analysis was performed with FACSCalibur cytometer (BD Biosciences, NJ, USA), and data were analyzed by FlowJo software (BD Biosciences, NJ, USA).
In vitro induction of mesenchymal lineage differentiation of isolated MSCs
Multilineage differentiation potential of isolated porcine MSCs was evaluated by culturing the second-passage MSCs (3×104 cells/cm2 in 35-mm dish) with osteogenic or adipogenic induction medium for 3 weeks, as previously described (Bosch et al., 2006). Briefly, for osteogenic induction, differentiation media were replaced every 3–4 days; for adipogenic differentiation, cells were first exposed to induction medium for 3 days and then cultured in maintenance medium for another 1 days. This alternating treatment was repeated three times to achieve full adipogenic differentiation. Von Kossa staining and Oil Red O staining were performed as described by Pittenger et al. (1999).
Somatic cell nuclear transfer
Cultured MSCs and fibroblasts at fifth passage were used as donor cells for SCNT. The SCNT experiments for both fibroblasts and MSCs were performed under the same conditions and following the protocol described by us (Li et al., 2013). Briefly, each batch of ≈500–600 matured porcine oocytes was randomly separated into two equal portions; one portion was used for SCNT with fibroblasts and the other portion was used for SCNT with the same donor pig's MSCs.
Porcine ovaries were purchased from The Guangzhou Tianhe slaughterhouse (Tianhe district, Guangzhou city, P.R. China). We obtained permission from this slaughterhouse to use the porcine ovaries for SCNT experiments in our study. Cumulus–oocyte complexes (COCs) were aspirated from the ovaries and matured in vitro for 42–44 h following the protocol described by Deng et al. (2011). Matured COCs were freed from cumulus cells by repeated pipetting in 0.1% hyaluronidase. Matured oocytes with a first polar body were selected for cloning. The oocyte was sucked firmly onto the holding pipette so that it did not move. The enucleation pipette was inserted through the zona pellucida, and the first polar body and adjacent cytoplasm, presumably containing all the chromosomes, were aspirated into the enucleation pipette. Then a single fibroblast cell or a MSC was microinjected into the perivitelline space of the oocytes. The oocyte–donor cell complexes were cultured in porcine zygote medium 3 (PZM3) (Yoshioka et al., 2002) at 39°C for 1.5 h and then activated to fuse in a medium containing 250 mM mannitol, 0.1 mM CaCl2·2H2O, 0.1 mM MgCl2·6H2O, 0.5 mM HEPES, and 0.01% polyvinyl alcohol (PVA), by two successive DC pulses at 1.2 kv/cm for 30 μs using an electrofusion instrument (model CF-150/B, BLS company, Budapest, Hungary). The activated cloned embryos were then cultured in PZM3 medium containing cytochalasin B (5 μg/mL) for 4 h. After the postactivation treatment, the reconstructed embryos were then cultured in PZM3 medium at 39°C, 5% CO2, 7% O2, 88% N2, and 100% humidity.
In vitro culture of reconstructed embryos
MSCs-embryos and Fs-embryos were cultured under the same conditions and following the same protocol described below. Activated reconstructed embryos were transferred into PZM3 medium and cultured at 39°C, 5% CO2, 7% O2, 88% N2, and 100% humidity for 168 h (7 days). The time of embryo activation was defined as 0 h. The fusion rate, cleavage rate, and blastocyst rate of cultured embryos was assessed at 1 h, 24 h, and 168 h, respectively. The total cell number of blastocysts was counted at 168 h by staining the embryos with 1 μg/mL Hoechst 33342 and viewing the cell nuclei under a fluorescence microscopy.
Embryo transfer, diagnosis of recipient pregnancy, and delivery of cloned piglets
Transfer of MSCs-embryos and Fs-embryos reconstructed with a same batch of oocytes to recipients were performed under the same conditions and following the protocol described by us (Li et al. 2013). Briefly, MSCs-embryos and Fs-embryos reconstructed with a same batch of oocytes were transferred to recipient sows (about 250 embryos/recipient) raised in the same swine farm and with similar physiological conditions. Activated reconstructed embryos were transferred into PZM3 medium and cultured at 39°C, 5% CO2, 7% O2, 88% N2, and 100% humidity for 20 h. The embryos were then loaded into a tube and kept in a portable incubator (Minitube) during transportation to the recipient farm. Estrus-synchronized LY (Landrace ♂×Yorkshire ♀) or YL (Yorkshire ♂×Landrace ♀) hybrid sows in parity 2–5 were used as embryo recipients. Within 12 h after recipient sows showed signs of estrus with a standing response to boars, they were anesthetized with ketamine and xylazine for induction and 3% of isoflurane for maintenance. One oviduct was exposed by surgery. The cloned embryos were put directly into the oviduct of the recipient using a syringe.
The pregnancy status of the recipient sows was monitored using an ultrasound equipped with a convex transducer at 1 month after embryo transfer. If spontaneous farrowing did not occur until gestation day 116, the recipients were injected with a prostaglandin analog (200 μg/recipient) and after about 24 h they delivered by vaginal birth under supervision or with assistance. The newborn cloned piglets were clinically examined to check if they have any abnormalities, such as macroglossia, cryptorchidism, ligament contracture, cleft palate, and testes hypertrophy. The number of delivered cloned piglets was recorded for comparison between groups.
DNA methylation analysis by bisulfite sequencing
Cultured passage-5 MSCs and fibroblasts from the same donor Duroc pig were trypsinized and pelleted, and DNA was extracted by using E.Z.N.A.® Tissue DNA Kit (Omega Bio-Tek). DNA was eluted into 200 μL of elution buffer, and the concentration was measured at A260 using NanoDrop 2000 (Thermo).
Approximately 500 ng of purified genomic DNA was treated with sodium bisulfite to convert all unmethylated cytosine into uracil by using an EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA) according to manufacturer's recommendations. Briefly, DNA was diluted into 20 μL of distilled water and then denatured at 98°for 10 min in 130 μL of the CT Conversion Reagent. Denatured DNA was incubated at 64°C for 2.5 h in the dark. Bisulfite-treated DNA was then desalted, purified, and diluted in 20 μL of M-Elution Buffer. Subsequently, 1–4 μL of converted DNA was used per run in bisulfite-specific PCR amplification.
Bisulfite-modified DNAs were amplified by PCR with previously described primers for short interspersed nuclear elements (SINEs) (Hyldig et al., 2011) and centromeric satellite sequences (Kang et al., 2001), and by nested PCR with previously reported primers for Oct4 DMR1 (Wu et al., 2009), Nanog promoter (Miyamoto et al., 2009), IGF2 DMR2 (Han et al., 2008), and H19 CTCF3 (Han et al., 2008). Nested PCRs were run using HotStarTaq plus DNA polymerase (Qiagen) with 25–30 cycles of the first amplification reaction and 45 cycles of the second amplification reaction. The PCR amplification of SINEs and satellite region were performed for 45 cycles. The amplified products were verified by electrophoresis on 3% agarose gels and gel-purified using an E.Z.N.A.® Gel Extraction Kit (Omega Bio-Tek). Purified PCR fragments were cloned into the TA cloning vector pTZ57R/T (Fermentas). Positive colonies were confirmed by colony PCR and sent for sequencing until at least 10 qualified sequences at each differentially methylated region (DMR) locus were obtained. Sequences were analyzed by local BiQ Analyzer software, and a bead diagram was plotted on the web site at http://biq-analyzer.bioinf.mpi-inf.mpg.de/tools/MethylationDiagrams/index.php
Transfection and selection of MSCs
The third-passage Yorkshire MSCs were cultured in 6-cm dishes until reaching 90% confluence. The cells were transfected with linearized ≈15 kb of plasmid pPSP-xynB (for plasmid structure, see Fig. 4A, below) by Lipofectamine (Invitrogen). At 24 h posttransfection, the cells were selected with 500 μg/mL G418 (Gibco) for 14 days. Surviving transgenic cell colonies were pooled and grown in dishes for 9 days. Transgenic MSCs at the sixth passage were used as donor cells for SCNT, as described above, to produce transgenic cloned pigs.
PCR, Southern blot and reverse transcription PCR analysis of transgenic MSCs and transgenic cloned pigs
For PCR analysis, genomic DNA was isolated from selected G418-resistant MSCs and the ear biopsy of transgenic cloned piglets. A set of primer P1+P2 (for location of primers, see Fig. 4A, below) was used to amplify a 567-bp fragment of the xynB transgene from the isolated genomic DNA by PCR. A 680-bp fragment of the reference gene β-actin also was amplified to examine the quality of isolated genomic DNA. The PCR amplification product was sequenced to confirm their sequence identity.
For Southern blot analysis, 10 μg of tail genomic DNA was restriction digested with SacI and separated by electrophoresis in 0.8% agarose gel. The DNA was then transferred to a nylon membrane (Amersham Pharmacia Biotech) by the capillary transfer method. During transfer, the DNA was nicked and denatured. The membrane was prehybridized overnight at 42°C and then hybridized with a 600-bp xynB transgene probe labeled with digoxigenin (DIG) by using a PCR DIG Probe Synthesis Kit (Roche). Hybridization and washing were performed with a DIG-High Prime DNA Labeling and Detection Starter Kit II (Roche). After hybridization, the membrane was incubated for 30 min in blocking solution and further incubated for 30 min in anti-DIG-AP antibody solution. The membrane was then exposed for 5–20 min after incubation with 1 mL of CSPD ready-to-use, and the picture was captured by the EC3 imaging system (UAP Company, CA, USA).
Two transgenic pigs were euthanized at the age of 1 year to analyze transgene expression. Various tissues were dissected, cut into tiny pieces, and frozen in liquid nitrogen. Total RNA was isolated by a RNA Extraction Kit (Qiagen) and then was reverse transcribed into cDNA, followed by DNase I treatment to eliminate any contamination of genomic DNA. A set of primer P1+P2 (for location of primers, see Fig. 6A, below) was used to amplify a 567-bp fragment of the xynB transgene from the cDNA by PCR. A 301-bp fragment of the reference gene glyceraldehyde 3-phosphate dehydrogenase(GAPDH) also was amplified to examine the quality cDNA. The PCR amplification product was sequenced to confirm their sequence identity.
Statistical analysis
Data were analyzed by SAS 9.2 program (SAS Institute, Cary, NC), and the two-samples t-test for means program was used for mean comparisons. Data were presented as mean±standard deviation (SD). The significant difference of means between two different groups was determined at P≤0.01.
Results
Characterization of cell-surface markers of isolated MSCs
The cell-surface antigen profile of isolated MSCs was examined by flow cytometer following immunofluorescence cell staining. The results (see Fig. 1) showed that the porcine MSCs isolated from bone marrow were positive for three MSC markers, including CD29 (β1-integrin), CD44 (hyaluronate receptor), and CD105 (endoglin), but negative for the hematopoietic cell marker CD45 (leukocyte common antigen) (Chen et al., 2009; Wu et al., 2007). This suggests that the cells isolated from bone marrow were typical MSCs instead of hematopoietic origin.
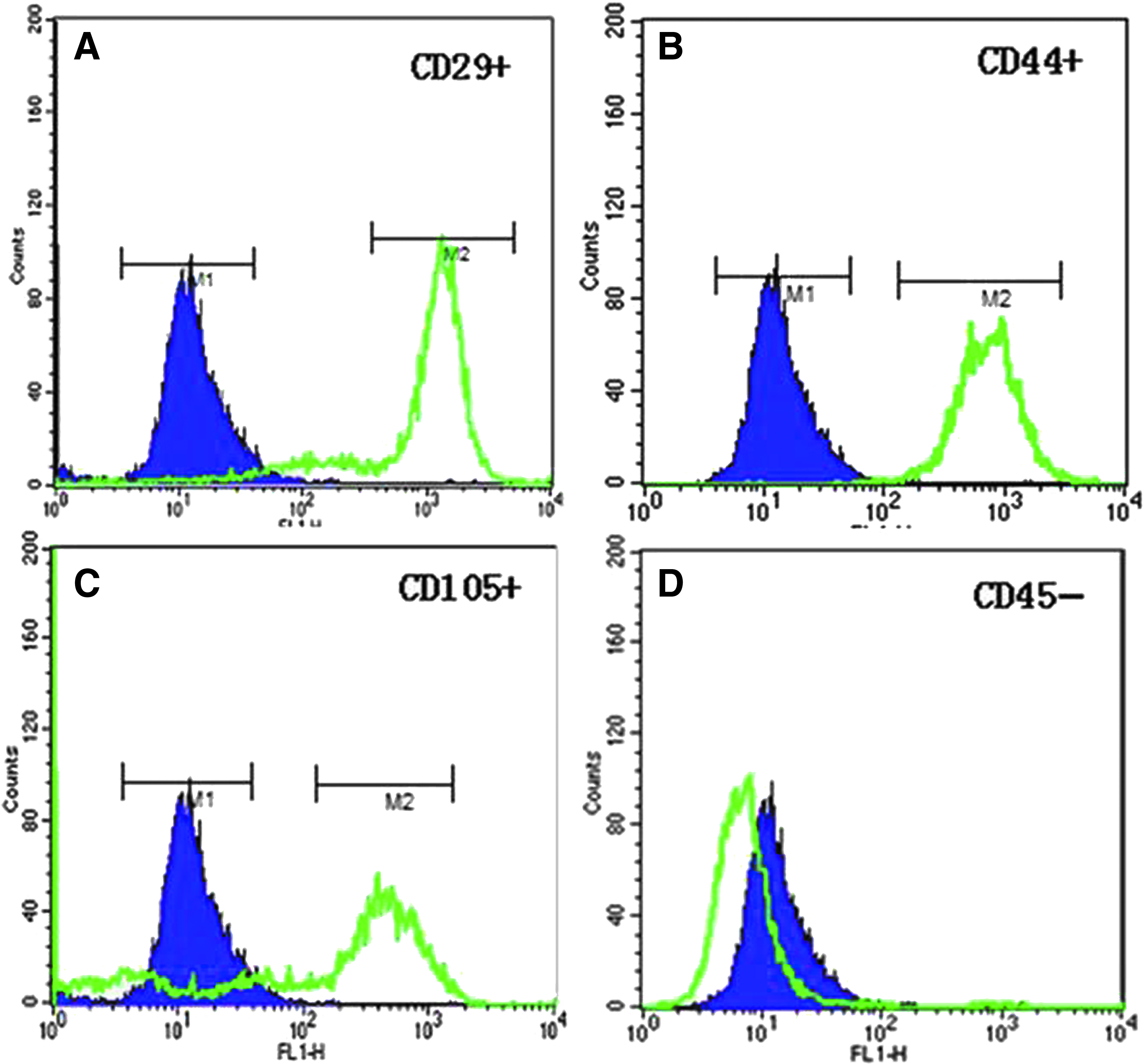
Characterization of cell-surface markers of isolated MSCs. Immunofluorescence-stained MSCs were analyzed by flow cytometry. The results are presented as histograms that show cell fluorescence intensity on the horizontal axis and cell frequency distribution on the vertical axis. In each histogram, the blue curve represents the distribution of cells incubated with the immunoglobulin isotype control, and the green curve represents the distribution of cells incubated with anti-CD29, anti-CD44, anti-CD105, or anti-CD45 primary antibody, and subsequently with green fluorescent dye-labeled secondary antibody. From a representative MSC line, 84.8%, 98.2%, and 60.0% of the total cells were positive for MSC markers CD29 (
In vitro mesenchymal lineage differentiation of isolated MSCs
The isolated MSCs appeared as single stretched or spindle-shaped cells in culture (Fig. 2A). They were differentiated into osteocyte-like cells with formation of extracellular mineralized matrix after exposure to osteogenic medium (Fig. 2B). Under adipogenic conditions, a lipid droplet that is typically seen in adipocytes was formed in differentiated MSCs (Fig. 2C). The ability of isolated MSCs to differentiate into various mesenchymal lineages suggests that these MSCs were multipotent.
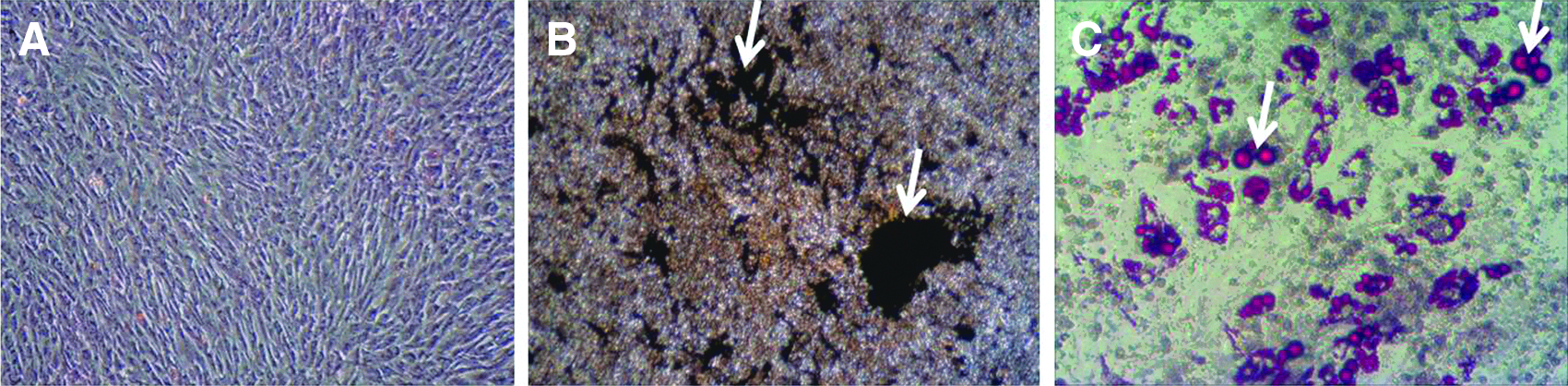
Osteogenic and adipogenic differentiation of isolated MSCs. (
Comparison of in vitro preimplantation developmental ability between Fs-embryos and MSCs-embryos
For each of the three investigated pig breeds, including Duroc, Landrace, and Wuzhishan, embryos cloned from MSCs and fibroblasts of a same donor pig have no significant (p>0.05) difference in fusion rate, cleavage rate, and blastocyst rate (Table 1). However, in each of studied swine breeds, the total cell number in blastocysts cloned with MSCs was significantly (p≤0.01) higher than that in blastocysts reconstructed from a same donor pig's fibroblasts (Table 1). For total embryos derived from three pig breeds, MSCs-embryos have 49.73% (28.0±4.7 vs. 18.7±5.1, p≤0.01) more total cells than Fs-embryos at the blastocyst stage (Table 1). Figure 3 shows a representative picture showing that MSCs-embryos have more total cells than Fs-embryos at the blastocyst stage.
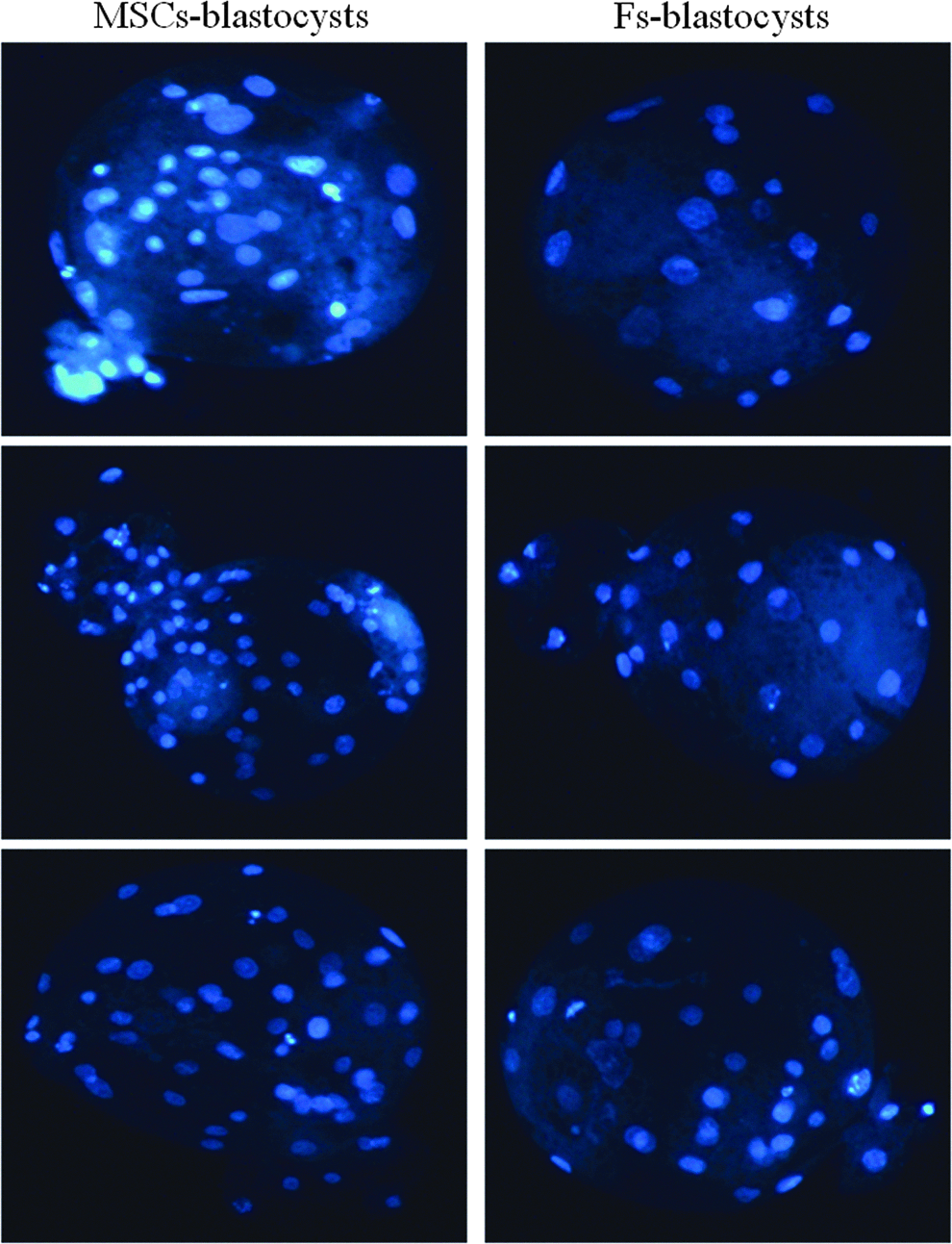
Comparison of total cell number between MSCs-blastocysts and Fs-blastocysts cloned from the same cell donor pig. Blastocysts were stained with Hoechst 33342, and total cells were counted under fluorescence microscopy. This representative picture shows that blastocysts reconstructed with MSCs (MSCs-blastocysts) have more total cells than blastocysts cloned from fibrobalsts (Fs-blastocysts).
Ear fibroblasts (Fs) and bone marrow mesenchymal stem cells (MSCs) were isolated from a Duroc (D), a Landrace (L), and a Wuzhishan (W, a Chinese local swine breed) piglet at the age of 3–5 days. Values of each breed were calculated based on the data obtained from three repeated experiments and presented as mean±standard deviation (SD). The two values of the same column and of the same cell donor pig are statistically different at p≤0.01 if they have different superscripts, but are not statistically different (p>0.05) if they have no superscript.
Comparison of in vivo full-term developmental rate between Fs-embryos and MSCs-embryos
Transfer of a total of 1221 embryos cloned with a Duroc pig's MSCs into five recipients at approximately the two-cell stage resulted in four pregnancies at 1 month after embryo transfer; one of the pigs had a miscarriage at 2 months of gestation, and the remaining three pregnant recipients maintained full-term pregnancy and gave birth of a total of 11 live healthy cloned piglets (Table 2). However, within the five surrogate sows that received a total of 1276 embryos cloned from a same Duroc pig's fibroblasts, only one was diagnosed as pregnant at 1 month after embryo transfer; yet abortion was found in this pregnant recipient at 56 days of pregnancy, and therefore no cloned offspring was born from the Fs-embryos group (Table 2). The pregnancy rate and farrowing rate of recipients and the birth rate of cloned piglets resulted from transfer of MSCs-embryos were higher than that of Fs-embryos group (Table 2). Figure 4 shows some of the piglets cloned from Duroc MSCs. The 11 cloned piglets delivered by the MSCs-embryos recipients were born alive and did not show visible defects, such as macroglossia, cryptorchidism, ligament contracture, cleft palate, and testes hypertrophy, which are commonly seen in cloned piglets. The birth weight of 11 piglets cloned from Duroc MSCs was between 0.6 kg and 0.9 kg, which was within the normal birth weight range (0.6–1.8 kg) of noncloned Duroc piglets born at the same pig farm.
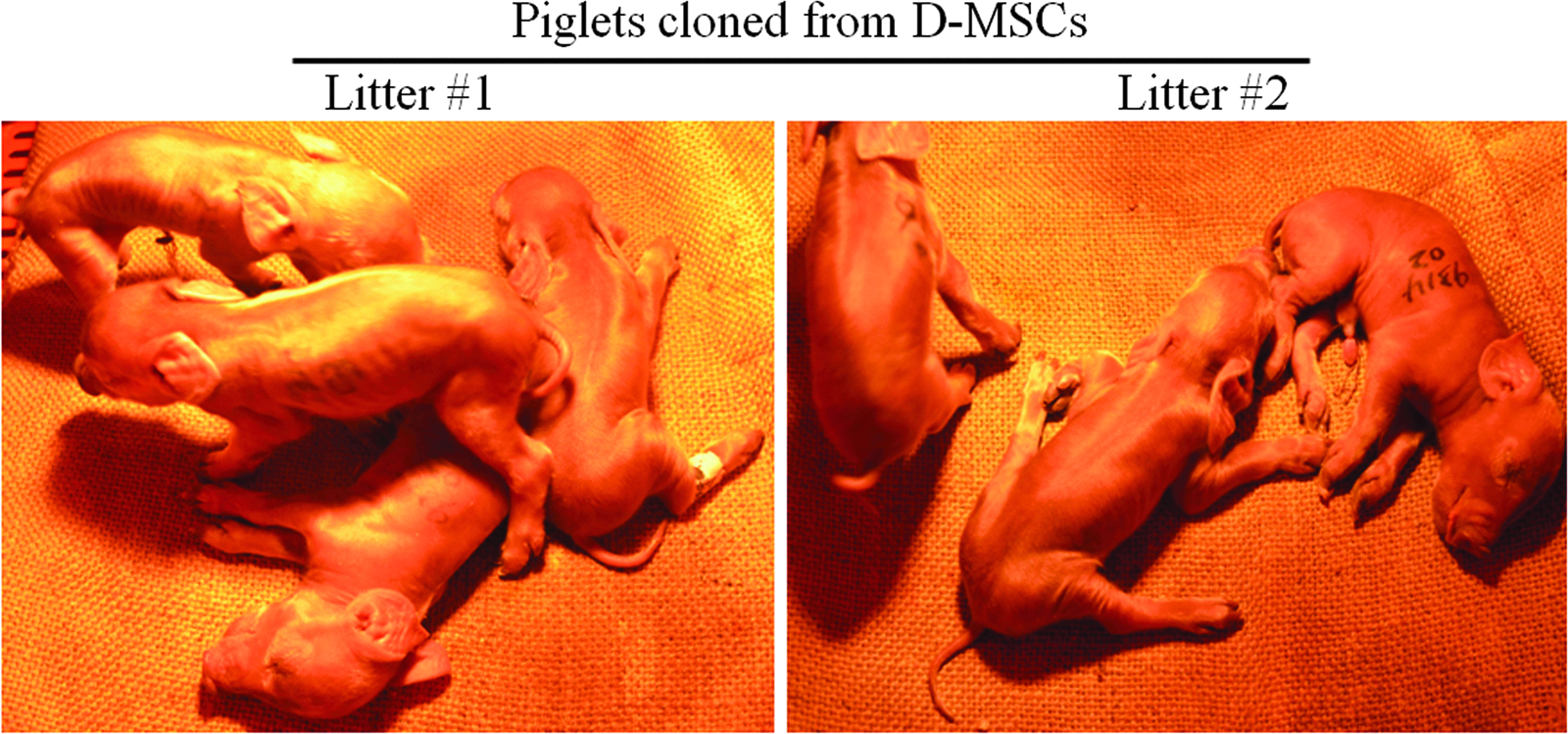
Piglets cloned from Duroc MSCs (D-MSCs).
Cloned embryos were transferred at approximately two-cell stage.
Pregnancy of recipients was examined at 1 month after embryo transfer.
Abortion in both of these two recipients was observed at about 2 months of gestation.
All these 11 piglets cloned from MSCs were born alive, had normal birth weight, and showed no visible defects.
Litter size of farrowed three recipients were four, three, and four, respectively.
LY (Landrace ♂×Yorkshire ♀) and YL (Yorkshire ♂×Landrace ♀) are hybrid sows used for embryo recipients.
Comparison of DNA methylation level of Oct4, Nanog, IGF2, H19, SINEs, and Satellite between Fs and MSCs
To examine whether the two donor cell types associated with distinct SCNT efficiency differ in DNA methylation pattern, we analyzed their DNA methylation degree in regions well representing two pluripotency genes, including Oct4 and Nanong, two imprinting genes, including IGF2 and H19, and two repetitive elements, including SINEs and Satellite. The results indicated that Fs and MSCs are similar in methylation level of Oct4 (69.4% vs. 69.4%, see Fig. 5A) and Nanog (21.7% vs. 20.0%, see Fig. 5B). However, methylation degrees of both IGF2 and H19 in Fs are higher than that in MSCs (72.0% vs. 67.0% and 46.5% vs. 28.3%, see Fig. 5C, D). Euchromatic SINEs are more methylated whereas heterochromatic satellite sequences are less methylated in Fs, as compared to that in MSCs (59.09% vs. 55.88% and 59.60% vs. 66.40%, see Fig. 5E, F).
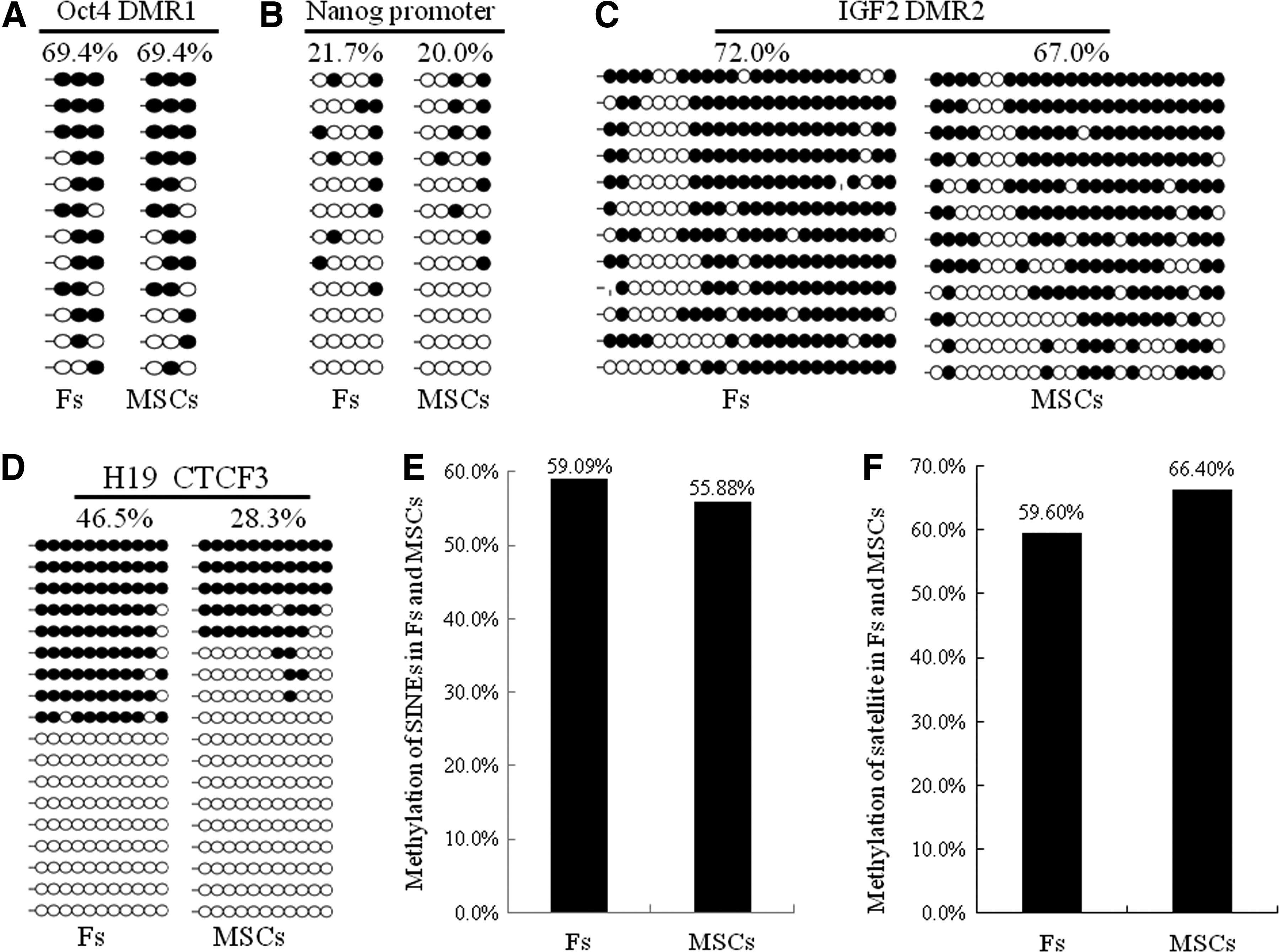
Comparison of DNA methylation levels of Oct4, Nanog, IGF2, H19, SINEs, and Satellite between Fs and MSCs isolated from the same Duroc pig. In
Production of transgenic cloned pigs using genetically modified MSCs as donor cells for SCNT
To further investigate whether MSCs could be used as donor cells for production of genetically modified pigs via SCNT, we transfected a Yorkshire pig's bone marrow MSCs with a plasmid harboring a salivary gland-specific promoter-driven fungal xylanase gene and a cytomegalovirus (CMV) promoter-controlled neomycin-resistant gene (Fig. 6A), and selected the cells with G418 for 2 weeks. The resulting G418-resistant transgenic MSCs (Fig. 6B, C) were pooled and used as donor cells to reconstruct 478 embryos by SCNT. Following the transfer of these cloned embryos at approximately two-cell stage into two surrogate sows, four live cloned piglets were born and two of them were identified as transgenic pigs carrying the xylanase transgene, which was specifically expressed in two salivary glands of transgenic pigs (Table 3 and Fig. 6D–H).
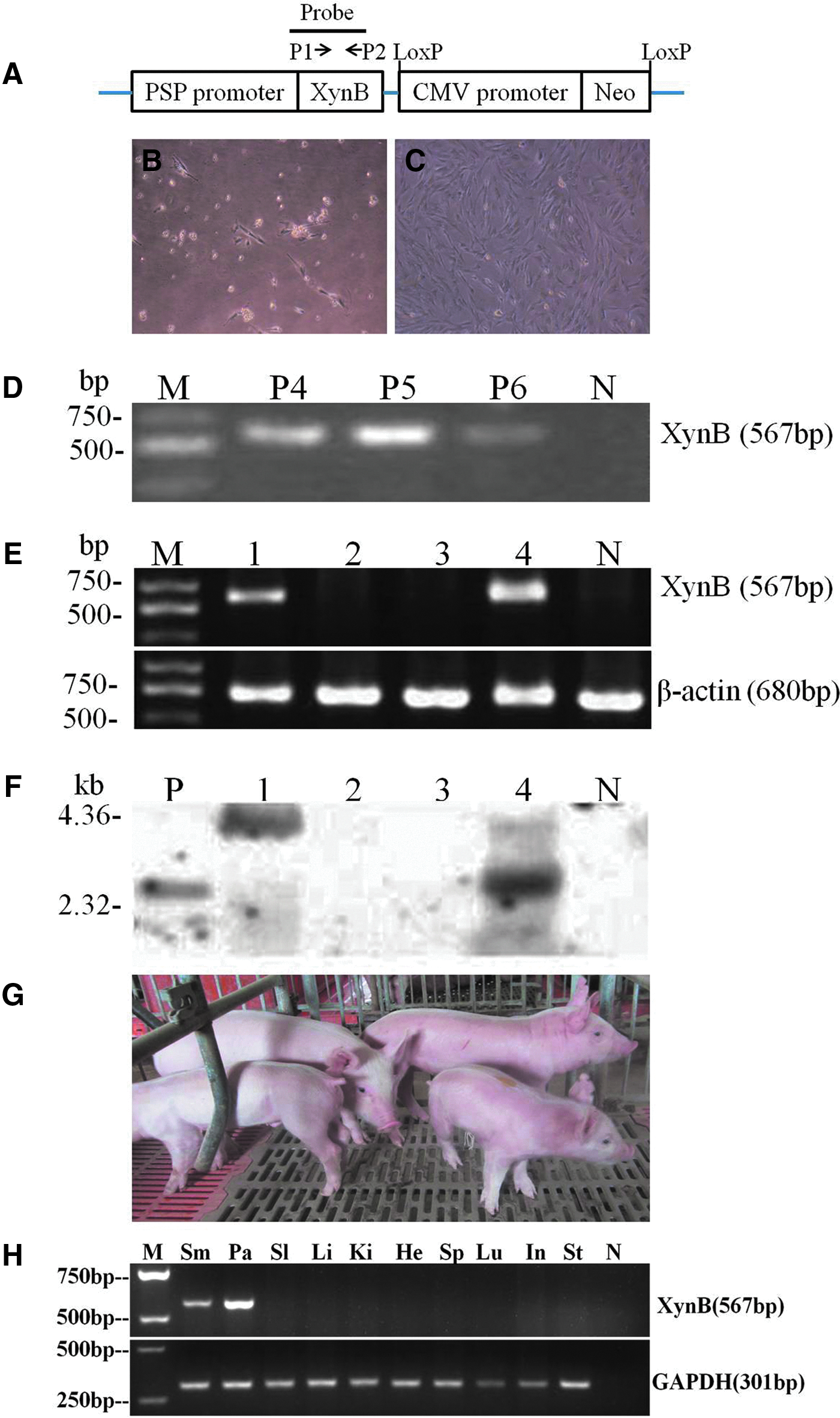
Production of transgenic cloned pigs using genetically modified MSCs as donor cells for SCNT. (
Cloned embryos were transferred at approximately two-cell stage.
All of these four piglets were born alive, had normal birth weights, and showed no visible defects; two of the four cloned piglets were identified as transgenic pigs carrying the xynB transgene.
Discussion
It has been hypothesized that after nuclear transfer the genome of undifferentiated or less differentiated cells, such as embryonic stem cells (ESCs) and adult stem cells, may be more easily reprogrammed to a pluripotent state by the recipient oocytes. In mice, studies already showed that embryos cloned from ESCs have enhanced full-term survival ability (Eggan et al., 2001; Rideout et al., 2000) compared to reconstructed embryos derived from somatic cells (Wakayama and Yanagimachi, 1999). However, because there are no porcine ESCs with proven pluripotency available now, MSCs, a type of multipotent adult stem cells, were employed as donor cells to improve the success rate of pig cloning. Although the porcine MSCs-embryos were demonstrated to have higher preimplantation developmental competence than Fs-embryos in in vitro culture (Colleoni et al., 2005; Faast et al., 2006; Jin et al., 2007; Kumar et al., 2007; Lee et al., 2010), their full-term developmental ability has not yet been compared with that of Fs-embryos. In this study, we showed that porcine MSCs-embryos have higher blastocyst total cell numbers as well as full-term developmental potential, and porcine MSCs can be reprogrammed to generate live transgenic cloned pigs following stable genetic modification.
In the present study, the bone marrow MSCs isolated from neonatal pigs not only expressed MSC-specific antigens on the cell surface, but also could be induced to differentiate toward osteogenic and adipogenic lineages. This multipotency of isolated MSCs indicates that they carry a nucleus with higher plasticity, which could be more efficiently remodeled to drive normal development of cloned embryos after being transferred to enucleated oocytes.
We observed in this study that at the blastocyst stage the cloned embryos derived from MSCs of three different pig breeds have 49.73% (28.0±4.7 vs. 18.7±5.1, p≤0.01) more total cells than Fs-embryos. A similar result was also found in some reported studies (Jin et al. 2007; Lee et al., 2010). The total cell number of preimplantation embryos was shown to be critical for the progression of normal embryonic development (Machaty et al., 1998). MSCs-embryos have a higher cell counts than Fs-embryos, suggesting they are of higher quality than Fs-embryos. However, no statistically significant difference in fusion rate, cleavage rate, and blastocyst rate was found between in vitro–cultured MSCs-embryos and Fs-embryos of three investigated swine breeds. This is not consistent with that seen in previous studies (Colleoni et al., 2005; Faast et al., 2006; Jin et al., 2007; Kumar et al., 2007; Lee et al., 2010), which all reported that the MSCs-embryos have a significantly (p≤0.05) higher blastocyst rate than Fs-embryos.
Although MSCs-embryos and Fs-embryos showed a similar fusion rate, cleavage rate, and blastocyst rate in preimplantation development, they were different in full-term developmental rate, as we demonstrated that the birth rate of live piglets cloned from MSCs was 0.90% (=11/1221=number of delivered live cloned piglets/number of transferred cloned embryos), which was significantly higher than the cloning efficiency associated with Fs (0%). However, cloned piglets have been obtained by some researchers with the efficiency of 1.3–2.4%, even though they used Fs as a donor cells (Kim et al., 2010; Zhao et al., 2009). Because cloned embryos were treated with drugs that can improve reprogramming prior to embryo transfer in the Zhao et al. study, and preselected high-quality oocytes were used for cloning in the Kim et al. study, these could be the main reasons causing the difference in cloning efficiency between our study and their studies. In addition, the difference in cloning efficiency between our study and their studies could also be caused by many aspects, such as difference in genetics (breed) and epigenetics of donor cells, source and quality of oocytes, procedure of nuclear transfer, procedure of embryo transfer, physiology of recipients, etc.
In our study, the cloning efficiency comparison between MSCs and Fs with the same genetics (isolated from a same donor pig) was conducted by using the same source of oocytes, under the same conditions and following the same procedure. Our result showed that the cloning efficiency associated with MSCs (0.9%) is significantly higher than that resulting from using Fs as donor cells for cloning (0%). Moreover, our study showed that all the cloned piglets derived from MSCs were born alive and did not exhibit any visible defects, which usually could be seen in piglets cloned from Fs (Li et al., 2013; Schmidt et al., 2010). This strongly suggests that MSCs can serve as suitable donor cells that could be more efficiently reprogrammed to produce cloned pigs through SCNT, as compared with fibroblasts. The enhancement of full-term developmental ability observed in MSCs-embryos probably was due to an increase of total cell number at the blastocyst stage, which enhanced subsequent postimplantation development. Lee et al. (2010) showed that use of MSCs as donor cells for cloning give rise to a birth rate of live cloned piglets at 0.76% (=4/523=number of delivered live cloned piglets/number of transferred total cloned embryos), which is close to that (0.90%=11/1221) observed in this study. Nevertheless, comparison of the birth rate of live cloned piglets between MSCs-embryos group and Fs-embryos group was not performed in the Lee et al. (2010) study.
Our results revealed that DNA methylation status of two pluripotency factors, Oct4 and Nanog, is very similar in porcine bone marrow MSCs and ear fibroblasts, even though these two cell types exhibited different nuclear reprogramming ability during SCNT. This is in accordance with the result reported by Streckfuss-Bomeke et al. (2012). Their study showed that human bone marrow MSCs and skin fibroblasts also have no significant difference in methylation level of Oct4 and Nanog genes, although MSCs showed a significantly higher reprogramming efficiency than fibroblasts when they were reprogrammed to generate human induced pluripotent cells (iPSCs).
Genome-wide DNA methylation analysis showed that human bone marrow MSCs and skin fibroblasts have different DNA methylation profiles on global promoter regions; CpG sites within the promoter of genes involved in regulation of development are hypomethylated in MSCs, in comparison to fibroblasts (Koch et al., 2011). This suggests that a lower methylation level of development-related genes is needed to maintain the multipotent capacity of MSCs. Therefore, it was not a surprise to see in our study that methylation degree of two imprinting genes, IGF2 and H19, both of which play important roles in development (Baker et al., 1993; Brunkow and Tilghman 1991; Leighton et al., 1995; Nezer et al., 1999; Van Laere et al., 2003), are lower in MSCs than that in fibroblasts.
Centromeric satellites represent the heterochromatic DNA regions (Kang et al., 2001), whereas SINEs are diffusely distributed in the euchromatic chromosomal regions of porcine genome (Thomsen and Miller, 1996). Like the methylation change pattern of global genomic DNA, both SINEs and Satellite are highly methylated in porcine gametes, especially in sperm, yet are quickly demethylated in porcine embryos following in vivo fertilization (Kang et al., 2001). This implies that a fast demethylation of heterochromatic satellites and euchromatic SINEs after fertilization is required for normal development of porcine embryos. If this is true, then donor cells with a lower methylation level of repeat sequences probably will be more easily demethylated during early development of cloned embryos, which is favorable for normal embryonic development. However, we found that in this study the MSCs carrying less methylated euchromatic SINEs but more methylated heterochromatic satellite sequences exhibited a higher nuclear reprogramming rate than fibroblasts during SCNT.
Our data indicate that differences in the DNA methylation profile could be related to the distinction in nuclear reprogramming efficiency between MSCs and fibroblasts during SCNT. Nevertheless, other types of epigenetic modification in nuclear donors could also affect SCNT efficiency, because the histone methylation pattern was reported to differ remarkably between MSCs-originated and fibroblasts-derived rabbit SCNT embryos (Brero et al., 2009).
The SCNT technique is not only commonly used to duplicate desired pigs but also is widely employed to create genetically modified pigs. Although we already showed that use of MSCs as donor cells can significantly improve pig SCNT efficiency, we still want to know whether the porcine MSCs could be used as donor cells to generate genetically engineered pigs by SCNT. The donor cells suitable for transgenesis SCNT should be adaptable to genetic manipulation as well as drug selection, and support full-term development of genetically modified cloned embryos after their nuclei were transferred to enucleated oocytes. In this study, we demonstrated that in vitro-cultured Yorkshire MSCs were amenable to transgene plasmid transfection and subsequent G418 selection, and surviving G418-resistant transgenic MSCs could be reprogrammed to generate live transgenic pigs following nuclear transfer. The cloning efficiency associated with Yorkshire MSCs was 0.84% (=4/478=number of born Yorkshire cloned pigs/number of transferred embryos), comparable with the cloning efficiency associated with Duroc MSCs (=0.9%, see Table 2), which are shown to be better than Fs when used as donor cells for cloning. The four cloned pigs or transgenic cloned pigs derived from Yorkshire MSCs were born alive and did not show visible defects, which usually could be found in piglets cloned from Fs (Li et al., 2013; Schmidt et al., 2010). In addition, use of Yorkshire MSCs as donor cells for production of transgenic pigs carrying the Xylanase gene resulted in a transgenic efficiency at 0.42% (=2/478=number of born transgenic cloned pigs/number of transferred embryos). Previously, we also used Pietrain fetal Fs cells to produce transgenic cloned pigs carrying the same Xylanase gene. The transgenic efficiency was 0.37% (data not reported yet), which is slightly lower than that associated with the Yorkshire MSCs showed in this study.
Two previous studies indicated that cultured MSCs can undergo transient as well as stable genetic modification by nonviral and viral vectors (Bosch et al., 2006; Colleoni et al., 2005), but production of genetically engineered cloned pigs from MSCs was not reported in these two studies. During the preparation of this manuscript, a research group reported that they used MSCs to generate gene-targeted pig models through the SCNT technique (Flisikowska et al., 2012; Leuchs et al., 2012). These two studies, together with our data, clearly show that MSCs can serve as donor cells to produce genetically modified pigs by SCNT.
In conclusion, we have demonstrated that porcine SCNT embryos reconstructed with bone marrow MSCs have enhanced survival ability in full-term development, as compared to embryos cloned from fibroblasts. The increase in the developmental ability of MSCs-embryos could be linked with the DNA methylation status of their nuclear donors. In addition, we showed that porcine bone marrow MSCs can be used as donor cells to produce genetically modified pigs by SCNT. These results suggest that bone marrow MSCs are an attractive donor cell type for both SCNT and transgenesis SCNT in pigs.
Footnotes
Acknowledgments
This study was supported by a grant from the National High Technology Research and Development Program of China (863 Program, grant number 2011AA100304), a grant from Department of Science and Technology of Guangdong (grant number 2011A020901001), and a grant from Guangdong Science and Technology Project (grant number 2011A020102003). We thank Xiaoling Huang for his help in preparation of the manuscript.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.