
Abstract
Abstract
Recurrent chromosomal alterations have been repeatedly reported in cultured human embryonic stem cells (hESCs). The effects of these alterations on the capability of pluripotent cells to differentiate and on growth potential of their specific differentiated derivatives remain unclear. Here, we report that the hESC lines HUES-7 and -9 carrying multiple chromosomal alterations produce in vitro mesenchymal stem cells (MSCs) that show progressive growth arrest and enter senescence after 15 and 16 passages, respectively. There was no difference in their proliferative potential when compared with bone marrow–derived MSCs. Array comparative genomic hybridization analysis (aCGH) of hESCs and their mesenchymal derivatives revealed no significant differences in chromosomal alterations, suggesting that genetically altered hESCs are not selected out during differentiation. Our findings indicate that genetically unstable hESCs maintain their capacity to differentiate in vitro into MSCs, which exhibit an in vitro growth pattern of normal MSCs and not that of transformed cells.
Introduction
S
Materials and Methods
hESC culture
The hESC lines HUES-7 and HUES-9 (obtained from Prof. Douglas Melton, Harvard Stem Cell Institute) were maintained on BD Matrigel™ hESC-qualified Matrix (BD Biosciences, Bedford, MA
In vitro differentiation of hESCs to MSCs
To induce mesenchymal differentiation, hESCs were cultured in suspension for 7 days in ultra-low-attachment culture dishes (Corning Life Sciences, Corning, NY, USA) for the formation of embryoid bodies (EBs). Then, approximately 70 EBs/well were plated onto 0.1% gelatin-coated six-well plates in the presence of human (h) MSC growth medium supplemented with 4 ng/mL basic fibroblast growth factor (bFGF; Invitrogen). Cells were cultured for up to 2 weeks to reach confluence and then detached using 0.05% trypsin–EDTA (Gibco BRL, Grand Island, NY, USA). Afterward, they were continually passaged into T25 flasks, until they took the appearance of a homogeneous fibroblastic morphology. Cells derived from primary cultures were defined as passage 0 (P0), and each cycle of reseeding of hESC-MSCs after trypsinization was considered to be one additional passage.
Bone marrow–derived MSCs
Bone marrow (BM) was harvested from the posterior iliac crest of two normal donors, aged 8 and 12 years old, for a related stem cell transplant after informed consent and approval from the Ethical Committee of the Aghia Sophia Children's Hospital (Athens, Greece). Mononuclear cells were isolated from 5 mL of donor BM and suspended in Dulbecco's modified Eagle medium (DMEM; Stem Cell Technologies), enriched with 10% autologous serum, and placed into a 75-cm2 flask (Corning Life Sciences, Corning, NY, USA) at a concentration of 150,000 cells/cm2. The flasks were incubated at 37°C in a humidified environment with 5% CO2. By 9–12 days, a homogeneous population of adherent fibroblastoid cells was present. At this time point, cells were detached with 0.05% trypsin–EDTA for 5 min at 37°C, counted, and subsequently replated in a ratio of 1:3. Cells derived from primary cultures of BM–mononuclear cells were defined as P0, and each subsequent cycle of reseeding of MSCs after trypsinization was considered to be one additional passage.
Surface antigen analysis
For fluorescence-activated cell sorting (FACS) analysis, single-cell suspensions of collected hESC-MSCs were analyzed using an Epics XL-MCL (Beckman Coulter, Inc.) flow cytometer. The following human-specific monoclonal antibodies were used: CD29-fluorescein isothiocyanate (FITC), CD90-phycoerythrin (PE), CD44-FITC, CD105-FITC, CD45-PC5, CD73-PE, CD19-FITC, CD31-PE, and HLA-DR-PE. Control staining with appropriate isotype-matched monoclonal antibodies was included in all FACS experiments.
Differentiation into osteogenic, adipogenic, and chondrogenic lineages
hESC-MSC differentiation into osteogenic, adipogenic, and chondrogenic lineages was achieved using established reported methodologies (Dominici et al., 2006).
Osteogenesis
To stimulate osteogenic differentiation, cells were plated at a density of 1.5×104 cells/cm2 in T25 flasks with a medium consisting of MesenCult® MSC Basal Medium, supplemented with 15% Osteogenic Stimulatory Supplement, 10-mM β-glycerophosphate, 0.01 μM dexamethasone, and 50 mg/mL ascorbic acid (Stem Cell Technologies). Cells were fed twice a week for a period ranging from 3 to 5 weeks.
Adipogenesis
For induction of adipogenesis, cells were plated for 2 weeks at a density of 4.0×104 cells/cm2 in T25 flasks with a medium consisting of MesenCult® MSC Basal Medium, supplemented with 10% MesenCult® Adipogenic Stimulatory Supplements (Stem Cell Technologies).
Chondrogenesis
For induction of chondrogenesis, cells were cultured as a high-density pellet culture (2.5×105 cells/pellet) in StemPro® Osteocyte/Chondrocyte Differentiation Basal Medium with 10% StemPro® Chondrogenesis Supplement (all from Gibco, BRL), for approximately 3 weeks.
Gene expression analysis by RT-PCR
Reverse transcription polymerase chain reaction (RT-PCR) was performed to analyze expression levels of pluripotency markers (OCT3/4, REX-1, SOX2, NANOG) on hESCs, hESC-MSCs, and BM-MSCs. Osteogenic, adipogenic, and chondrogenic differentiation were indicated by the expression of alkaline phosphatase (ALP), lipoprotein lipase (LPL), and collagen type II (COL2A1), respectively. Results were compared to expression levels of housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
aCGH analysis
Agilent Human Genome CGH+SNP 4×180K or 1×244K microarrays were used in this study (Agilent Technologies, Santa Clara, CA, USA, www.agilent.com) to investigate the genomic stabilility of the original hESCs (HUES-7 and HUES-9) and the hESC-derived MSCs. The 244K platform is composed of >236,000 60-mer oligonucleotide probes for the mapped genes or unique DNA sequences with an average spatial resolution of 7.9–8.9 kb [annotated against National Center for Biotechnology Information (NCBI) build 36, hg18]. The 4×180K CGH+SNP (SurePrint G3 ISCA CGH+SNP) platform is composed of 110,712 (CGH)+59,647 (SNP) 60-mer oligonucleotide probes with 25.3 kb overall median probe spacing [5 kb in International Standards for Cytogenomics Array (ISCA) regions] (annotated against NCBI build 37, hg19). For the location of genes in the deleted/duplicated genomic segments, the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/) and the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) were used.
Results
Generating MSCs from hESC lines
To investigate the differentiation potential of genetically altered hESCs, we induced MSC differentiation from hESCs using EB formation as previously described (Brown et al., 2009). When EBs were plated on gelatinized tissue culture plates in the absence of feeder cells and in serum-free medium that was supplemented with serum replacement medium, a homogeneous culture of fibroblast-like cells was generated within 2 weeks. The cultures had a fibroblastic cellular morphology that resembles adult MSCs (Fig. 1A). Surface antigen profiling of MSCs derived from hESCs by FACS analysis revealed a surface antigen profile that is qualitatively similar to that defined for BM-MSCs, that is, CD90+, CD29+, CD44+, CD73+, and CD105+, while they were CD45−, CD31−, HLA-DR−, and CD19− (Fig. 1B).
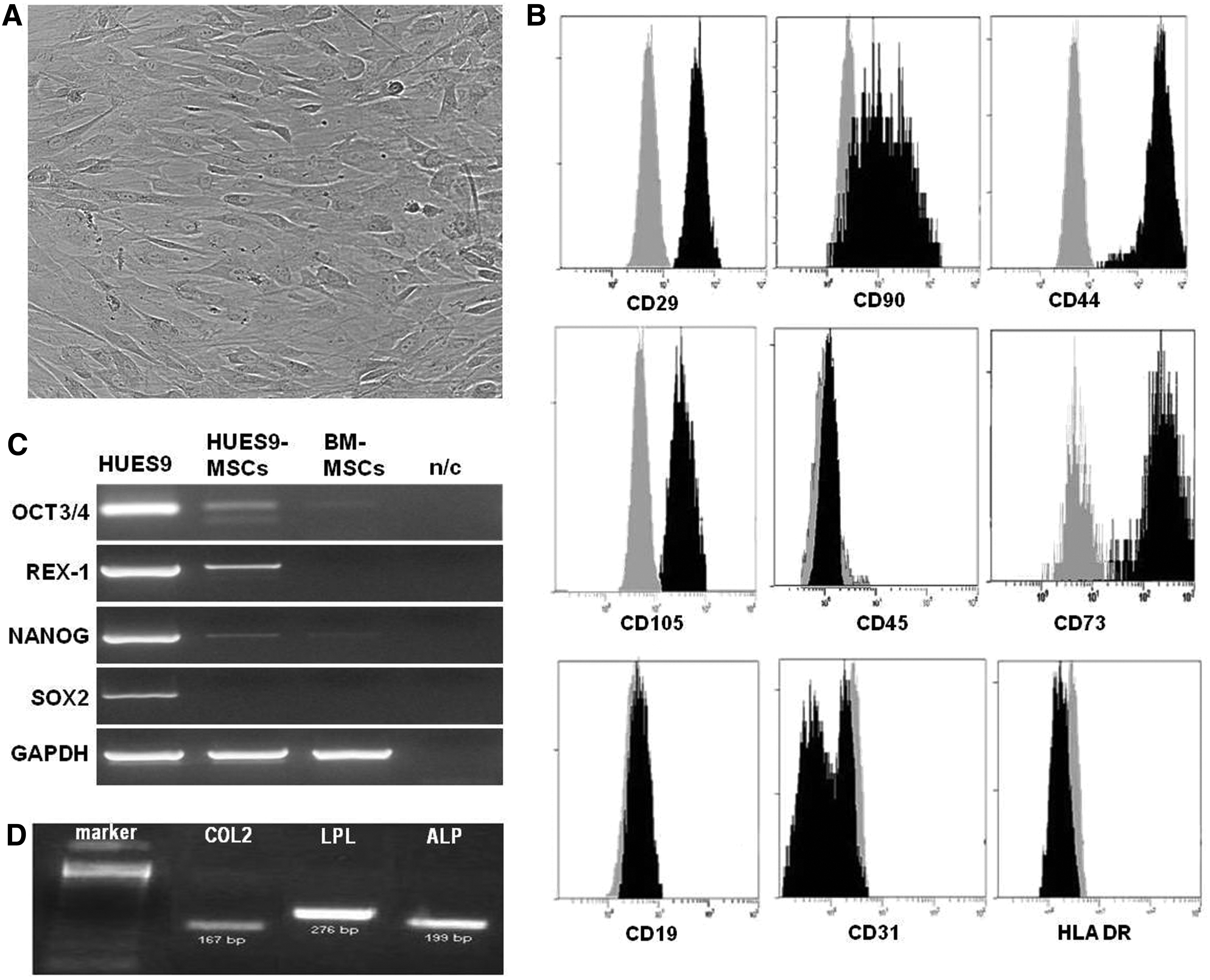
Characterization of MSCs derived from hESC lines HUES-9 and HUES-7. (
We performed RT-PCR to analyze the expression of OCT3/4, REX-1, NANOG, and SOX2 in hESC-MSCs. Although we have found some expression of OCT3/4, NANOG, and REX-1 in hESC-MSCs, it was clearly weaker in comparison with the expression of the pluripotency markers in undifferentiated embryonic cells. A low expression of OCT3/4 and NANOG was also detected in BM-MSCs (Fig. 1C).
The differentiation potential of hESC-derived MSCs was tested using standard differentiation conditions for adipogenesis, chondrogenesis, and osteogenesis. MSCs derived from HUES-7 and HUES-9 cells treated with osteogenic medium underwent a change in their morphology from spindle shaped to cuboidal, and they formed large nodules after 18 days of induction. MSCs cultured in adipocytic differentiation medium formed lipid vacuoles 14 days after induction. Chondrogenic, adipogenic, and osteogenic differentiation were confirmed by COL2A1, LPL, and ALP expression, respectively (Fig. 1D).
The mesenchymal derivatives of hESCs were trypsinized when confluent and split 1:3. HUES-7- and HUES-9-derived mesenchymal cell cultures had been maintained in continuous culture for 15 and 16 passages, respectively, at 1:3 split every week before they entered into senescence. The average population doubling times of HUES7-MSCs and HUES9-MSCs were 4.6 and 4.4 days, respectively.
BM-MSCs were grown in parallel to directly compare their proliferative potential to that of mesenchymal hESC derivatives. They grew as adherent spindle-shaped fibroblastic like cells and were positive for CD105, CD90, CD73, CD29, CD44, and negative for CD45, CD31, CD19, and HLA-DR. In addition, BM-MSCs exhibited differentiation potential in vitro toward the osteoblastic, adipogenic, and chondrogenic lineages. They exhibited quite similar proliferative potential to that of hESCs-mesenchymal derivatives
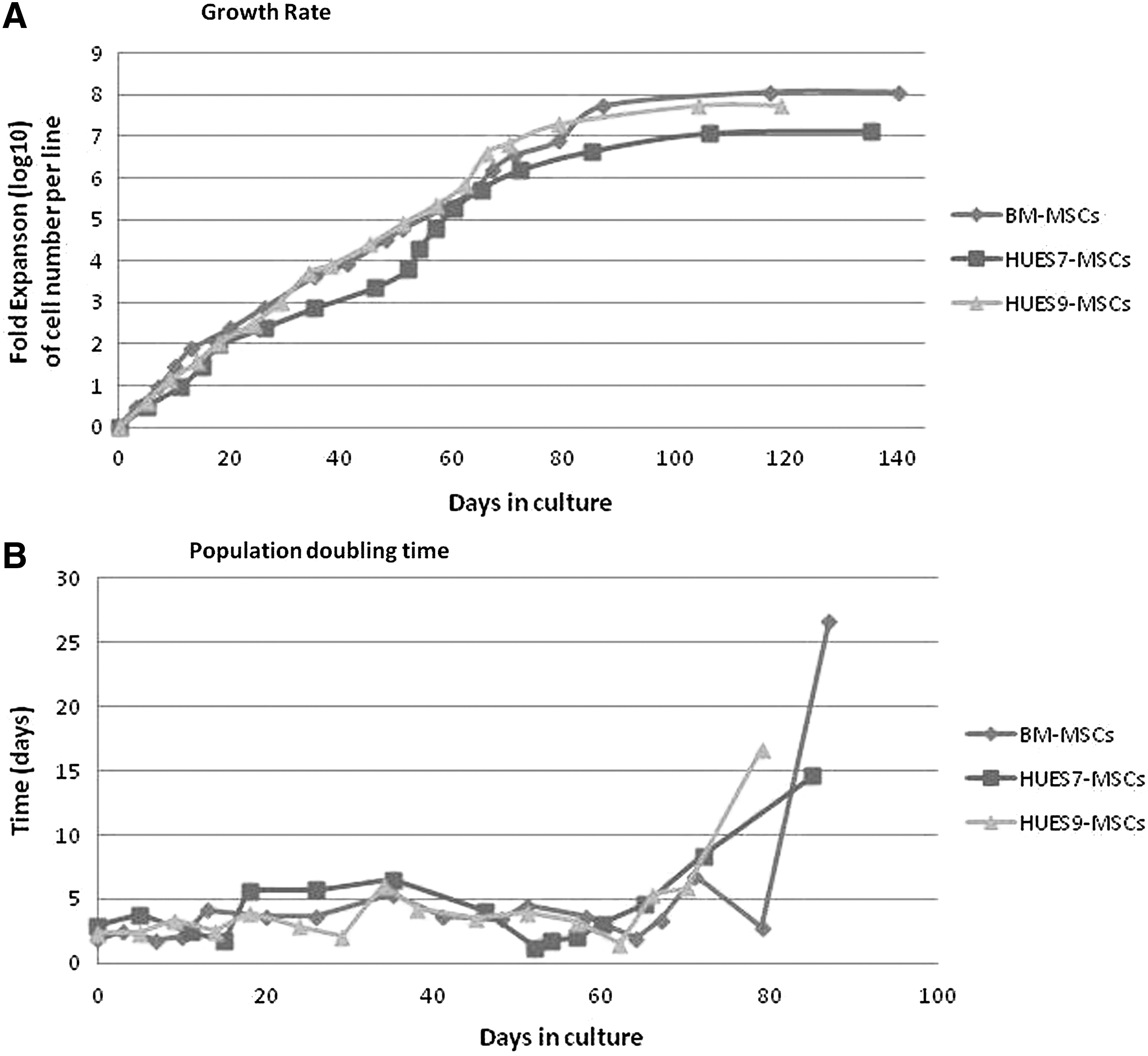
Growth characteristics and comparison of the proliferative potential of the mesenchymal hESC derivatives to that of BM-derived cells. (
Genetic alterations of hESCs at their undifferentiated stage and of mesenchymal derivatives
By using aCGH, we analyzed samples obtained from feeder cell free cultures of HUES-7 and HUES-9 hESCs lines at passages 29 and 51, respectively. At the undifferentiated stage, the HUES-9 line exhibited chromosomal aberrations, including duplications at 1p34.3–p34.2, 3p22.1, and 20q11.21–q11.21, whereas whole chromosome 12 and 17 gains at 12p13.33–p11.1, 12q12–q24.33, 15q11.2, 17p13.3–p11.2, 17q11.1–q25.3, and a deletion of 10q11.22 were detected in HUES-7 (Fig. 3 and Table 1). To find out whether genetically altered hESCs lose their capacity for differentiation or whether differentiation can be a selective process for or against cell populations carrying specific aberrations, we also tested mesenchymal derivatives from HUES-7 and -9 cell lines at passage 4, using the same aCGH procedure. In both cell lines, the chromosomal structural aberrations that were present in the undifferentiated hESC populations were maintained in their mesenchymal derivatives, with slight differences in size, whereas no new aberrations arose during the course of differentiation (Table 1).
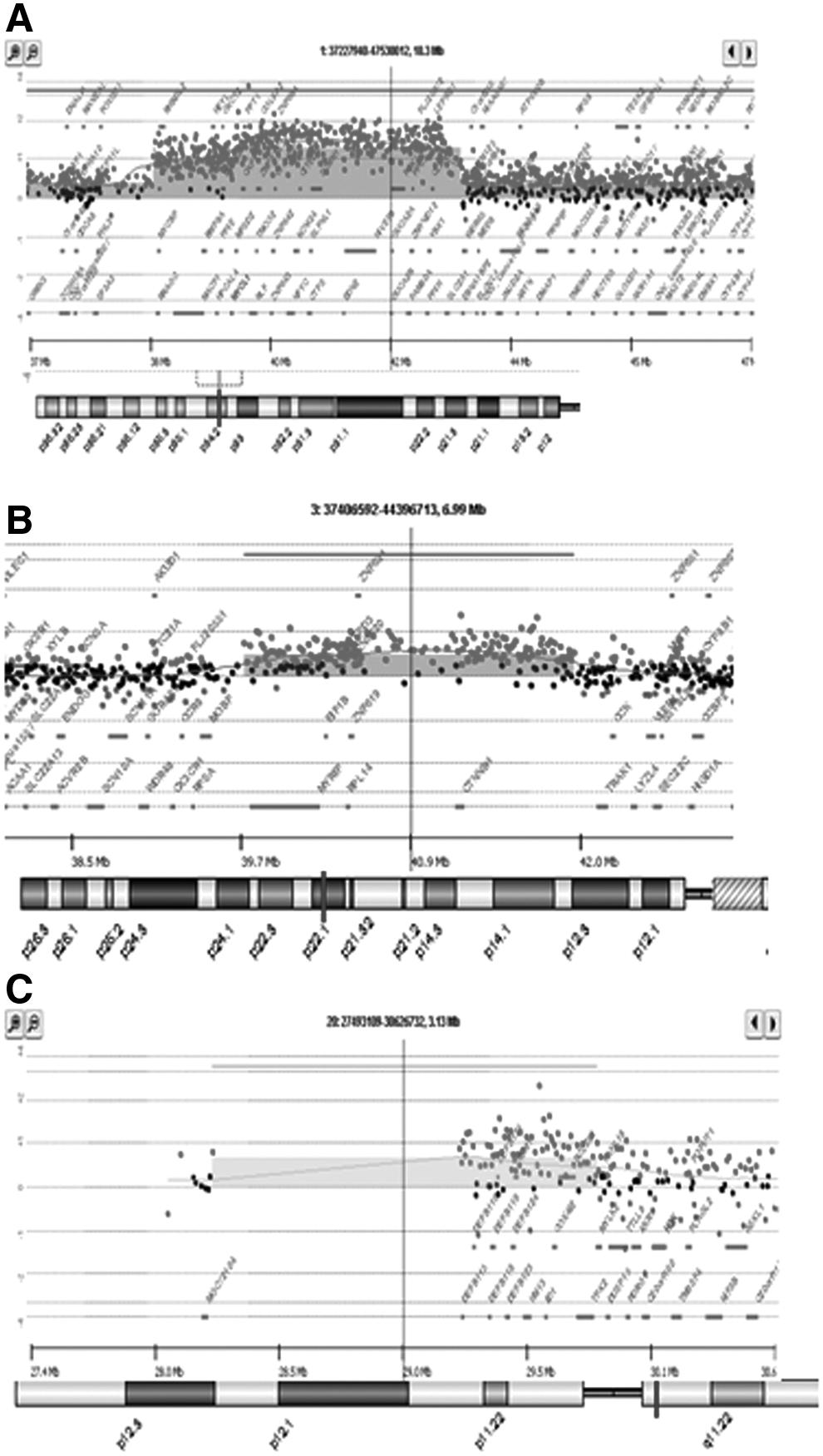
A1×244K array-CGH profile of HUES-9 cells showing: (
CNVs, copy number variations; hESCs, human embryonic stem cells; hMSCs, human mesenchymal stem cells; CGH, comparative genomic hybridization; SNP, single-nucleotide polymorphism; DUP, gain; DEL, loss; Mb, megabases; Kb, kilobases.
Discussion
The fact that genetic and epigenetic alterations in hESCs occur during prolonged culture has raised serious doubts about their utility for cell therapy, disease modeling, and drug development (Lund et al., 2012; Nguyen et al., 2013). Recently, the International Stem Cell Initiative after an analysis of 125 hESC lines reported that most cell lines remained karyotypically normal, but there was a progressive tendency to acquire alterations on prolonged culture, commonly affecting chromosomes 1, 12, 17, and 20. They postulated that abnormalities are twice as likely to be present at high passages (33%) than in the early passages (14%). The chromosome instability of both hESC lines (HUES-7 and HUES-9) after prolonged culture in our hands was probably introduced during the culture adaptation period and gave a group of cells with the specific chromosomal aberrations selective advantage. Especially, gains of chromosomes 12 and 17 affect regions that contain genes implicated in apoptosis and differentiation. Furthermore, the cell cycle–related gene and key pluripotency factor NANOG is located in chromosome 12p (Draper et al., 2004). A minimal amplicon in chromosome 20q11.21 was also identified to probably be related to increased survival of hESCs in vitro. Three genes are located in this region—BCL2L1, ID1, HM13. Of these, BCL2L1 is considered to be a strong candidate for driving culture adaptation and conferring growth advantage to hESCs, and ID1 encodes a protein that binds helix–loop–helix transcription factors to inhibit lineage commitment and affect cell growth (International Stem Cell Initiative et al., 2011).

4X180K CGH+SNP array-CGH profile of HUES-7 cells showing: (
Most studies have focused on genetic alterations detected in hESCs at their undifferentiated stage and have not paid much attention to their derivatives, which are the final cell products solely applicable in clinical protocols. Therefore, it is essential to investigate whether such genetic instability may influence functional properties of the pluripotent hESCs, including their capacity for differentiation. Moreover, if these aberrations persist through in vitro differentiation, they may provide an increased growth potential to differentiated cells, making them unsafe for clinical applications. There have been few recent investigations of chromosomal abnormalities in their differentiated derivatives showing a rather altered differentiation potential. The first evidence suggesting impaired differentiation was the reduced dependence on bFGF of hESCs to remain undifferentiated. Withdrawal of bFGF from the culture induces spontaneous differentiation with loss of stage-specific embryonic antigen-3 (SSEA3) expression in karyotypically normal hESCs but not in hESCs carrying chromosomal alterations (Herszfeld et al., 2006; Werbowetski-Ogilvie et al., 2009). Furthermore, neural derivatives of hESCs with 20q11.21 amplification continue to carry this alteration and show a more immature phenotype compared with neural cells derived from normal hESCs (Werbowetski-Ogilvie et al., 2009). Similar results were obtained by Fazeli and colleagues, who reported differences in the expression of several genes in EBs derived from genetically altered hESCs compared to karyotypically normal ones (Fazeli et al., 2011). In another recent study, however, the authors using a differentiation model of hESCs into endothelial cells showed that the ability of CHA3-hESCs with trisomy of chromosome 12 was unaffected when compared to karyotypically normal CHA3-hESCs. Transplanted karyotypically abnormal CHA3-hESCs endothelial derivatives formed a tumor-like tissue in two out of seven mice tested (Moon et al., 2011). In line with these in vivo results, a recent study reported that transplantation of neural progenitors derived from genetically altered hESCs developed into neuroectodermal tumors (Werbowetski-Ogilvie et al., 2012).
Using a differentiation culture method of hESCs through EB formation, we obtained MSCs that exhibited the same phenotype as those derived from BM and possess the capability to further differentiate to osteocytes, chondrocytes, and adipocytes as BM-MSCs do. Assessing the status of genetic aberrations in undifferentiated hESCs and in their mesenchymal derivatives obtained from late passages, we found that the same aberrations were maintained, whereas no new aberrations were observed (Table 1). Comparing their in vitro proliferative potential with parallel cultures of BM-derived MSCs, we found no significant difference in terms of doubling time and number of passages before they enter into senescence. Most reports indicate that genetically stable hESC-derived MSCs can be grown in culture for about 20–25 passages (Olivier et al., 2006).
There are no studies investigating the in vitro proliferative potential of differentiated cells derived from genetically unstable hESCs. However, Varela and colleagues showed that neural derivatives from normal hESCs acquired a 1q defect and could be propagated for more than 50 passages without entering senescence (Varela et al., 2012). Regarding MSCs, a recent study on both hESC-derived and somatic hMSCs has shown that these cells upon ex vivo expansion become genetically unstable by 4% (Ben-David et al., 2011). In addition, genetic alterations of clinical-grade BM-derived MSCs acquired during in vitro expansion were not associated with a selective growth advantage in vitro and conferred a growth disadvantage to altered cells, likely linked to DNA damage–associated senescence (Tarte et al., 2010). Therefore, we can hypothesize that genetic alterations affecting specific pathways like p53 or p16 may cause senescence bypass in culture (Whibley et al., 2010), whereas the contrary effect can be exhibited by aberrations affecting DNA repair systems (Rocha et al., 2013).
Our results show that genetically altered hESCs are capable for mesenchymal differentiation, with their mesenchymal derivatives also carrying the same chromosomal alterations. Moreover, this genetic instability detected in mesenchymal derivatives of hESCs was not associated with a selective in vitro growth advantage. We conclude that any abnormality in human pluripotent cells must be followed during in vitro differentiation as well as the behavior both in vitro and in vivo of their differentiated derivatives.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.