
Abstract
Abstract
The generation of induced pluripotent stem cells (iPSCs) from somatic cells by expressing ectopic reprogramming transcriptional factors such as Oct3/4, Sox2, Klf4, c-Myc, and Nanog is one of the cutting-edge discoveries in stem cell and cancer research. This discovery has raised several safety issues regarding the use of iPSC technology for human disease research. Tumorigenesis is the major obstacle observed for iPSC-mediated transplantation therapy. Recently, a new method to generate human iPSCs either by a chemical method or by direct delivery of reprogramming factors has become a promising approach for future customized cell therapy of human disorders. These reprogramming transcriptional factors play critical roles in diverse cellular functions such as transactivation, cellular proliferation, differentiation, apoptosis, and tumorigenesis. Posttranslational modifications (PTMs) (phosphorylation, ubiquitination, acetylation, sumoylation, and so on) of these proteins act as a regulatory signal to control protein activity, expression, and stability in a wide variety of cellular processes. We attempt to summarize the accumulated evidence to address the role of PTMs of Oct3/4, Sox2, Klf4, c-Myc, and Nanog in regulating their biological functions. This review allows us to understand the importance of PTMs and their application in developing an efficient and safe reprogramming method without cancer development for cell therapy. Finally, we discuss the importance of PTMs of reprogramming factors in tumor pathogenesis.
Introduction
E
In 2006, Takahashi and Yamanaka showed that retrovirus-mediated overexpression of a combination of four transcription factors, such as octamer 3/4 (Oct3/4), SRY box-containing gene 2 (Sox2), Krüppel-like factor 4 (Klf4), and c-Myc, so-called reprogramming transcription factors, was sufficient to reprogram mouse adult fibroblasts to an embryonic-like state (Takahashi and Yamanaka, 2006). The final reprogrammed cells were called induced pluripotent stem cells (iPSCs). The following year, the same group generated iPSCs from human dermal fibroblasts (Takahashi et al., 2007). Two other groups succeeded in generating iPSCs from human somatic cells using slightly different combinations of genes, including Oct3/4, Sox2, Nanog, and LIN28A (LIN28) (Park et al., 2008; Yu et al., 2007).
The generation of iPSCs avoids the ethical and political issues of using embryos and oocytes. The fact that iPSCs can also be generated from patients made it more reliable and beneficial than using ESCs for the study or treatment of human-related diseases. There are several hurdles that need to be overcome to develop a safe iPSC technology for clinical trials. First, the use of lentiviral or retroviral vectors to enable integration of transgene often leads to mutations within the host genome. Second, the reactivation of transcriptionally silent proviruses during iPSC generation could lead to oncogenesis (Hacein-Bey-Abina et al., 2003a, b). Adenoviral or episomal vectors that mediate transient expression without genomic integration of reprogramming factors are not completely free from chromosomal disruption and show a very low frequency of iPSC generation (Stadtfeld et al., 2008; Yamanaka, 2009; Zhou and Freed, 2009).
These reprogramming factors are highly regulated posttranscriptionally at the levels of mRNA stability, translation, and protein stability. Several different types of posttranslational modifications (PTMs) have been found on Oct3/4, Klf4, Sox2, c-Myc, and Nanog, including phosphorylation, ubiquitination, acetylation, and sumoylation (Baltus et al., 2009; Brehm et al., 1997; Chen et al., 2005; Evans et al., 2007; Hann, 2006; Moretto-Zita et al., 2010; Phanstiel et al., 2011; Ramakrishna et al., 2011; Wei et al., 2007; Williamson and Whetton, 2011; Xu et al., 2004), which are summarized in Table 1. PTMs of reprogramming proteins play significant roles in directing the cell fate determination of stem cells. The functional significance of PTMs on reprogramming factors is known to regulate their biological activities and protein stability. The phosphorylation of Oct3/4, Sox2, c-Myc, and Nanog alters their biological functions and modulates their transactivation activities (Bernier-Villamor et al., 2002; Hann, 2006; Jeong et al., 2010; Kang et al., 2009; Moretto-Zita et al., 2010). Ubiquitination of Oct3/4, Sox2, Klf4, c-Myc, and Nanog is responsible for regulating their half-life and subsequent protein stability (Chen et al., 2005; Hann, 2006; Hu and Wan, 2011; Jeong et al., 2010; Moretto-Zita et al., 2010 ; Ramakrishna et al., 2011; Xu et al., 2009; Xu et al., 2004). Acetylation of Sox2 and Klf4 is known to regulate their transcriptional activity (Jeong et al., 2010; Yu et al., 2011; Zhang et al., 2010). Additionally, ubiquitin-related modifier (SUMO) modification, known as sumoylation, was reported to enhance Oct3/4 stability, DNA binding, and transcriptional activity (Wei et al., 2007). Sumoylation of Klf4 resulted in increased transactivation activity (Du et al., 2010). Many studies that showed the effect of PTMs of the reprogramming factors on various biological functions and protein stability are summarized in Figure 1.
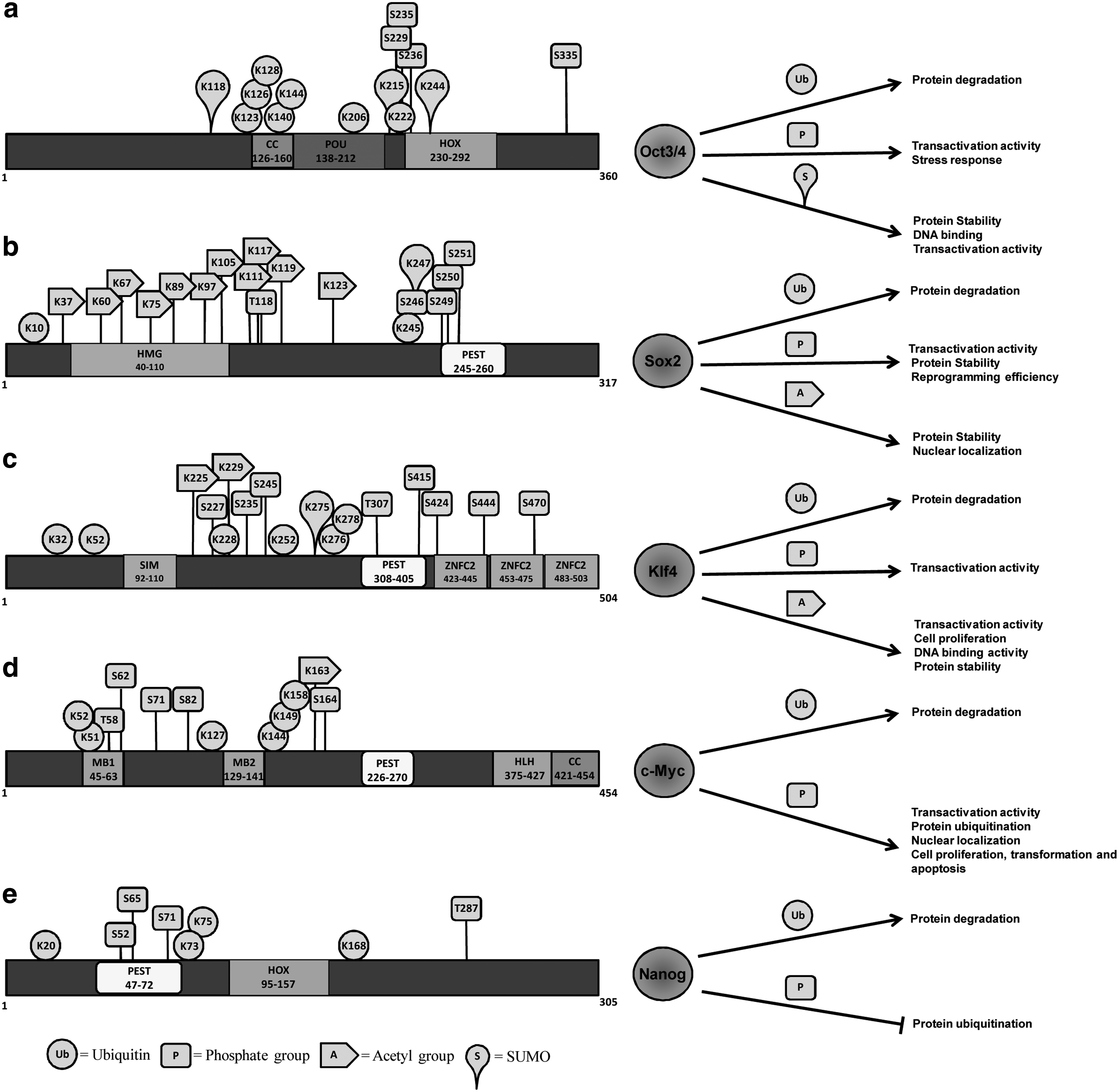
Schematic diagram of the human Oct3/4 (
N/D, not determined.
This review focuses on the role of PTMs of reprogramming transcriptional factors such as Oct3/4, Sox2, Klf4, c-Myc, and Nanog in regulation of proteolysis, transcriptional, and biological functions. Furthermore, we have reviewed the link between tumor pathogenesis and PTMs of defined reprogramming transcriptional factors.
PTMs of Oct3/4
The POU transcription factor, Oct3/4, plays a key role in ESC pluripotency and generation of iPSCs (Okamoto et al., 1990; Okita et al., 2007; Rosner et al., 1990). Oct3/4 expression occurs almost exclusively during the four- to eight-cell stage and ceases as cells undergo differentiation into multiple lineages (Brandenberger et al., 2004; Rosner et al., 1990; Scholer et al., 1990). The regulation of Oct3/4 expression and activity is very essential for maintaining the pluripotent state of ESCs. Apart from various factors affecting the expression level of Oct3/4, PTMs on Oct3/4 are also one of the main criteria in regulating its protein expression.
Oct3/4 undergoes rapid protein degradation and exhibits a relatively short half-life of about 90 min (Saxe et al., 2009). Ubiquitination of Oct3/4 is facilitated through the direct binding of the E3 ubiquitin ligase (Wwp2; WWP2) in mouse and human ESCs (Xu et al., 2004). Wwp2-induced ubiquitination of Oct3/4 negatively regulates transcription activities of both endogenous and overexpressed Oct3/4 in ESCs (Xu et al., 2004). Wwp2 catalyzes Lys-63 polyubiquitination of Oct3/4 and facilitates bringing these ubiquitinated Oct3/4 proteins to the 26S proteasome (Liao and Jin, 2010). Interestingly, Wwp2 could repress its own ubiquitin ligase activity by undergoing homodimerization at higher dosages and also mediate Oct3/4 ubiquitination during differentiation of ESCs (Liao and Jin, 2010). Thus, preventing ubiquitination of Oct3/4 might improve in maintaining ESC pluripotency.
Wei and colleagues reported that the stability of Oct3/4, DNA binding, and transactivation activity are enhanced by sumoylation (Wei et al., 2007). Oct3/4 showed three putative sumoylation sites at Lys-118, Lys-215, and Lys-244. Of these, Lys-118 is a critical one for Oct3/4 stability. Sumoylation of Oct3/4 increases protein stability and ability to activate transcription on both octamer sequences in a monomer manner or to PORE (palindromic Oct factor recognition element) and MORE (more PORE) sequences in alternate dimer conformations (Wei et al., 2007).
PIASy, an E3 SUMO ligase, interacts with Oct3/4 (Tolkunova et al., 2007) and functions as a capable transcriptional repressor of Oct3/4, but the activity is not associated with its native E3 SUMO ligase activity (Bernier-Villamor et al., 2002; Tatham et al., 2001). The SUMO-modifying enzymes PIASy, PIAS1, and PIAS3 showed association with Oct3/4 and modified its subnuclear localization in cell type–specific fashion, but the relocalization of Oct3/4 by PIASy is associated with repression of Oct3/4-mediated transcriptional activation, indicating its importance in mammalian development (Tolkunova et al., 2007).
Kang and colleagues analyzed the role of conserved phosphorylation target DNA-binding domain residues in Oct1 and Oct3/4 in regulation of binding selectivity (Kang et al., 2009). Oct3/4 showed responses to cellular stress signals by selectively altering the affinity for complex binding sites in vitro. A mutation at phosphorylation site Ser-335 on Oct3/4 shows alteration in binding selectivity for complex, indicating that the stress responsiveness of Oct3/4 operates through modification of phosphorylation sites (Kang et al., 2009). Saxe et al. have identified and reported that a putative protein kinase A (PKA) phosphorylation site at S229 lies within the POU domain of Oct3/4 (Saxe et al., 2009). A mutation of this phosphorylation site abolishes its transactivation activity on the PORE reporter, but is able to transactivate other Oct3/4 reporters. Furthermore, activators of PKA signaling substantially upregulated the expression level of Oct3/4 protein along with the upregulation of transcription of specific PORE genes. Furthermore, P38 MAP kinase, which functions downstream of PKA, mediates this phosphorylation and regulates Oct3/4 transactivation (Saxe et al., 2009). Although, transactivation activity of Oct3/4 is regulated by phosphorylation, its importance is yet to be tested in maintaining pluripotency and iPSC generation. Recently, O-linked N-acetylglucosamine (O-GlcNAc) has received far more attention due to its critical regulatory role in ESC pluripotency. Silencing of O-GlcNAc transferase (OGT), which is responsible for the addition of O-GlcNAc to targeted proteins, inhibits the pluripotency of ESCs, whereas increase in the O-GlcNAc levels perturbs normal ESC differentiation (Jang et al., 2012). The O-GlyNAcylation of Oct3/4 at Thr-228 regulates its transcriptional activity, resulting in the induction of several pluripotency-related genes, such as Klf2, Klf5, Nr5a2, Tbx, and Tcl1 (Jang et al., 2012). The O-GlyNAcylation of Oct3/4 is responsible for the cell fate determination of stem cells. Blocking O-GlyNAcylation of Oct3/4 stops its self-renewal efficiency in mouse ESCs, as well as the capability of generating iPSCs, indicating that O-GlyNAcylation of Oct3/4 is critical for maintaining pluripotency (Jang et al., 2012). Collectively, PTMs on Oct3/4 play a key role in regulating its transcriptional activity and protein stability. The roles of PTMs on the conserved domains of human Oct3/4 are summarized in Figure 1.
PTMs of Sox2
Sox2 is a member of large family of proteins characterized by their structure and sequence similarity to the Sry (sex-determining region of chromosome Y) protein. Along with Oct3/4, Klf4, and c-Myc, Sox2 was reported that it has the ability to re-establish pluripotency in terminally differentiated cells, reprogramming them to iPSCs (Takahashi and Yamanaka, 2006).
A phosphoproteome study in human ESCs by two independent groups revealed that the Sox2 protein contain four phosphorylation sites at Ser-246, Ser-249, Ser-250, and Ser-251 (Swaney et al., 2009; Van Hoof et al., 2009). The functional consequence of this serine phosphorylation induces sumoylation of Sox2 at Lys247 (Van Hoof et al., 2009), which in turn results in the inhibition of Sox2 DNA-binding activity (Tsuruzoe et al., 2006). The Akt-mediated Sox2 phosphorylation site Thr-118 near the high-mobility group (HMG) domain enhances Sox2 protein stability by suppressing the ubiquitin-mediated protein degradation. Furthermore, phosphorylation of Sox2 at Thr-118 has a significant role in mESCs differentiation and reprogramming of mouse embryonic fibroblasts (MEFs) into iPSCs, indicating its regulatory function during somatic cell reprogramming (Jeong et al., 2010).
p300/CBP mediates the acetylation of Sox2 protein in ESCs within the HMG DNA-binding domain (Baltus et al., 2009). Mimicking acetylation at K75 enhances Sox2 interaction with the nuclear export machinery, suggesting that the acetylation at K75 promotes Sox2 nuclear export, while blocking acetylation at K75 maintains Sox2 in the nucleus and sustains its target gene expression. Additionally, increased global acetylation levels in ESCs with deacetylase inhibitor treatment induce Sox2 ubiquitination, proteasome degradation, and abrogation of its target gene expression. This suggests that acetylation of Sox2 is critical for maintaining the protein level and ESC pluripotency (Baltus et al., 2009). However, on the basis of the fact that the usage of small molecules such as histone deacetylase (HDAC) inhibitors and DNA methyltransferase inhibitors showed improved mouse iPSC generation, further investigation on the association between acetylation of Sox2 and iPSC generation is necessary (Huangfu et al., 2008; Shi et al., 2008).
Initially, Sox2 protein was identified as an O-GlcNAcylated protein by high-throughput analysis for O-GlcNAc-glycosylated proteins in rat brain (Khidekel et al., 2004). Later, three major O-GlyNAcylation sites for Sox2 were identified at positions S248, T258, and S259 (Jang et al., 2012; Myers et al., 2011). The mutations on O-GlyNAcylation sites on Sox2 did not show much significant reduction in reprogramming efficiency, but the efficiency was reduced to a half when compared to wild-type Sox2 in mouse cells (Jang et al., 2012). Thus, O-GlcNAcylation of Sox2 is essential for somatic cell reprogramming. The conserved domains of human Sox2 and the role of PTMs are summarized in Figure 1.
PTMs of Klf4
Klf4 is a transcription factor that plays a key role in regulating cell proliferation and differentiation in several tissues (Nandan and Yang, 2009; Rowland and Peeper, 2006; Yoshida et al., 2008). Klf4 is one of the four defined transcription factors that is sufficient to produce pluripotent stem cells from normal fibroblast cells (Takahashi and Yamanaka, 2006). Recently, Klf4 was identified as an upstream regulator of a large feed-forward loop that contains Oct3/4, Sox2, c-Myc, and Nanog (Kim et al., 2008), suggesting an existence of a transcriptional hierarchy within the four reprogramming factors with both autoregulatory and feed-forward regulation.
Lys-225 and Lys-229 of endogenous Klf4 are acetylated by p300/CBP (Evans et al., 2007). Perturbation of these acetylation sites decreases the ability of Klf4 to activate target genes, indicating the importance of acetylation for Klf4-mediated transactivation. Furthermore, K225 and K229 of Klf4 are important for inducing p21CIP1/WAF1 and intestinal alkaline phosphatase (IAP), and are responsible for Klf4-mediated inhibition of cellular proliferation. Klf4 was found to modulate histone H4 acetylation differentially at the promoters of its target genes, whereas expression of Klf4 and HDAC3 together leads to the repression of the cyclin B1 gene (Evans et al., 2007). Meng et al. showed that all-trans retinoic acid (ATRA) increases Klf4 acetylation and its DNA-binding activity (Meng et al., 2009). ATRA-mediated HDAC2 phosphorylation at Ser-424 through the JNK (c-Jun NH2-terminal kinases) signaling pathway inhibits the association between HDAC2 and Klf4, causing subsequent increased Klf4 acetylation levels in vascular smooth muscle cell (VSMCs) (Meng et al., 2009). Zhang et al. showed that ATRA-induced mitofusin 2 (mfn-2) expression is dependent on Klf4 in VSMCs (Zhang et al., 2010). Klf4 upregulates mfn-2 expression by binding to the mfn-2 promoter. ATRA also induces Klf4 acetylation by increasing the interaction between Klf4 and p300, which in turn depends on Klf4 phosphorylation via activation of JNK and p38 MAPK signaling. Thus, acetylation of Klf4 increases its ability to transactivate the mfn-2 promoter (Zhang et al., 2010). ATRA promotes Klf4 acetylation by inducing Klf4 phosphorylation via JNK- and p38-dependent pathways, and acetylated Klf4 activates the SM22α promoter, whereas platelet-derived growth factor with two B chains (PDGF-BB) promotes Klf4 deacetylation by inducing Klf4 dephosphorylation via the ERK- and PI3K/Akt-dependent pathway, leading to suppression of the SM22α promoter. Thus, Klf4 shows different effects on the regulation of SMC marker genes in an acetylation-dependent manner (Yu et al., 2011).
Depending on its interacting partners, Klf4 functions as an activator or repressor of transcription for several genes either in cancer or normal cells. Variations in KLF4 protein levels during serum stimulation were associated with proteasomal degradation (Chen et al., 2005). KLF4 undergoes rapid protein degradation and exhibits a relatively short half-life of about 120 min (Chen et al., 2005). Thus, the investigation on the role of PTMs on Klf4 in maintaining stem cell pluripotency and generation of iPSCs is essential. The conserved domains of human Klf4 and the role of PTMs are summarized in Figure 1.
PTMs of c-Myc
c-Myc is a reprogramming inducer involved in direct activation of pluripotent marker genes and maintaining the pluripotent state in mouse ESCs (Takahashi and Yamanaka, 2006). c-Myc exhibits its biological functions by regulating the expression of target genes that are involved in cellular proliferation. Apart from transcriptional regulation of c-Myc expression, it has been reported that c-Myc expression is also regulated posttranscriptionally at the level of mRNA stability, translation, and protein stability (Hann, 2006). Several PTMs for c-Myc, such as phosphorylation, ubiquitination, and O-linked glycosylation, have been reported to regulate its biological functions (Hann, 2006).
Phosphorylation of c-Myc was shown to regulate several biological activities, including the regulation of c-Myc ubiquitination and proteolysis (Hann, 2006). Among several phosphorylation sites on c-Myc, Thr-58 and Ser-62 phosphorylation sites are important due to their presence in the conserved Myc Box I (MBI), a frequently mutated region in c-Myc proteins (Gupta et al., 1993; Lutterbach and Hann, 1994; Smith-Sorensen et al., 1996). Glycogen synthase kinase 3– (GSK-3)-mediated phosphorylation of c-Myc alters subnuclear localization of c-Myc (Gregory et al., 2003).
Later, several groups showed the biological significance of Thr-58 phosphorylation of c-Myc on cell proliferation, transformation, and apoptosis (Henriksson et al., 1993). The loss of Thr-58 phosphorylation showed increased B cell lymphomagenesis by inhibiting apoptosis (Hemann et al., 2005). Thus, Thr-58 phosphorylation of c-Myc may play a critical role in inducing apoptosis without affecting cell proliferation in cancer cells.
However, like Thr-58 phosphorylation, Ser-62 and Ser-71 were also shown to regulate c-Myc biological function. The extracellular signal–regulated kinase (ERK) has been shown to phosphorylate c-Myc in vitro (Lutterbach and Hann, 1994; Pulverer et al., 1994; Saksela et al., 1992). Serum stimulation of quiescent cells results in Ser-62 phosphorylation of c-Myc (Lutterbach and Hann, 1994; Sears et al., 2000). Oxidative stress showed a significant increase in Ser-62 phosphorylation in an ERK-dependent manner (Benassi et al., 2006), whereas studies by another group showed that overexpression of ERK in cells had no effect on Ser-62 phosphorylation (Lutterbach and Hann, 1994). Watnick et al. demonstrated that the sequential activation of PI3K, Rho, and Rho kinase (ROCK) increases Ser-62 phosphorylation in Ras-expressing cells (Watnick et al., 2003).
Phosphorylation of c-Myc plays an important role in regulating its transcriptional activity and also modulates target gene regulation. For example, small T antigen enhanced c-Myc phosphorylation to cause increased transactivation of the E2F2 promoter (Yeh et al., 2004). During the G2/M cell cycle phase, enhanced Ser-62 phosphorylation levels increase EMS-mediated promoter activity (Gupta et al., 1993). ERK-dependent phosphorylation of Ser-62 upon oxidative stress leads to increased c-Myc recruitment to the γ-glutamylcysteine synthetase (γ-GCS) promoter (Benassi et al., 2006). Similarly, oxidative stress–induced Ser-62 phosphorylation is found to enhance c-Myc recruitment to the human telomere reverse transcriptase (hTERT) promoter and reduces recruitment to cyclin D2 and cyclin B (Benassi et al., 2006). c-Myc phosphorylation represses an antiangiogenic factor thrombospondin-1 via sequential activation of PI3K, Rho, and ROCK (Watnick et al., 2003). Enhanced Ser-62 or Ser-71 phosphorylation is responsible for Bim regulation, which is involved in the regulation of apoptosis (Hemann et al., 2005). Ser-62, Ser-71, and Thr-58 phosphorylations suppress the expression of the proapoptotic BH3-only protein and Bim that are involved in apoptosis (Hemann et al., 2005). Taken together, c-Myc phosphorylations have shown to be critical for the transcriptional activity of c-Myc and also regulate the expression of several target genes in cancer cells or normal cells. However, the biological function of c-Myc phosphorylation might provide us valuable information to screen potential target genes that improve iPSC generation.
c-Myc is an unstable protein exhibiting a half-life of about 20–30 min regulated by the ubiquitin-proteasome pathway (Hann and Eisenman, 1984). The regions between amino acids 45 and 63 of MBI and between 126 to 144 amino acids of MBII are degrons (degradation signals) responsible for c-Myc proteolysis (Flinn et al., 1998). The amino-terminal region of c-Myc encoding 128 amino acids contains Myc degron signals for proteolysis (Salghetti et al., 1999). The MBI and MBII of c-Myc serve as binding regions for several ubiquitin ligases to regulate proteolysis instead of direct signaling for protein degradation (Gregory and Hann, 2000). Additionally, a PEST sequence (peptide sequence that is rich in proline [P], glutamic acid [E], serine [S], and threonine [T]) between amino acids 226 and 270 is responsible for rapid c-Myc degradation, but does not show any effect on c-Myc ubiquitination (Gregory and Hann, 2000).
S-phase kinase–associated protein (Skp) 2, an F-box protein in the ubiquitin ligase complex, significantly enhances the ubiquitination status of c-Myc (Kim et al., 2003). Additionally, Skp2-mediated ubiquitination of c-Myc has also been shown to increase transcriptional activity (Kim et al., 2003). The interaction between Skp2 and c-Myc leads to diminished c-Myc protein levels regulating c-Myc's cellular function by enhancing c-Myc–induced S phase entry (von der Lehr et al., 2003). In contrast, Fbw7, a component of the SCF ubiquitin ligase complex, regulates c-Myc proteolysis and transcriptional activity in a dose-dependent manner. The inhibition of GSK3 prevents Fbw7-mediated c-Myc proteolysis, suggesting that Fbw7-driven c-Myc turnover requires phosphorylation of c-Myc on Thr-58 by GSK-3 (Welcker et al., 2004). Another group showed that JNK interacts with c-Myc and promotes c-Myc ubiquitination and degradation (Alarcon-Vargas and Ronai, 2004). Taken together, c-Myc ubiquitination is a crucial regulatory mechanism involved in cell differentiation. Blocking the interaction between c-Myc and its specific E3 ubiquitin ligases might prevent its protein degradation and subsequently improve iPSC generation. The conserved domains of human c-Myc and the role of PTMs are summarized in Figure 1.
PTMs of Nanog
Nanog is a novel pluripotent cell-specific gene whose expression is restricted to ESCs and plays a key role in regulating pluripotency, maintaining the pluripotent epiblast, and preventing differentiation into primitive endoderm (Chambers et al., 2003; Mitsui et al., 2003). Reduction in the level of Nanog results in an increased tendency toward cell differentiation (Chambers et al., 2007; Hatano et al., 2005; Ivanova et al., 2006), suggesting the importance of Nanog expression level in maintaining ESC pluripotency.
The transcriptional regulation of Nanog at its proximal promoter containing an Oct3/4 and a Sox2 motif is responsible for recapitulating the desired amount of Nanog expression in pluripotent and nonpluripotent cells (Kuroda et al., 2005; Pan and Thomson, 2007; Rodda et al., 2005). FoxD3 was found to bind and activate the Nanog promoter through an ESC-specific enhancer localized −270 bp upstream of the transcription start site (Pan et al., 2006). Apart from transcriptional initiation and attenuation, the expression level and stability of Nanog protein are regulated by posttranscriptional mechanisms. Nanog was reported to be phosphorylated (Yates and Chambers, 2005), but the functional consequence of this posttranslational modification was not clearly understood. Nanog undergoes phosphorylation at Ser/Thr-Pro motifs in ESCs (Moretto-Zita et al., 2010). Phosphorylation of Nanog at four putative sites at Ser-52, Ser-5, and Ser-71, and Thr-287 promotes the interaction between Nanog and the prolyl isomerase Pin1, leading to increased stability and activity of Nanog by inhibiting its ubiquitination (Moretto-Zita et al., 2010). In contrast, mutating Nanog's phosphorylation sites abolished its interaction with Pin1, causing impaired self-renewal and decreased protein stability. Thus, Pin1 activity was shown to be critical for stabilizing Nanog protein by suppressing ubiquitination in ESCs and teratoma formation of ESCs in severe combined immunodeficient (SCID) mice (Moretto-Zita et al., 2010).
Recently, we demonstrated that Nanog undergoes ubiquitination both at Lys-48– and Lys-63–branched polyubiquitin chains. Nanog contains a PEST motif sequence from amino acids 47 to 72 at its amino-terminal region, which has been shown to target the protein for ubiquitination (Ramakrishna et al., 2011). In contrast, a PEST motif sequence-deletion mutation showed less ubiquitination, leading to Nanog protein stabilization. This indicates that the PEST motif is a signaling factor for Nanog proteolysis. Nanog has a short half-life of about 120 min in human ESCs. Treatment of human ESCs with proteasome inhibitor MG132 increased Nanog protein stability and extended its half-life (Ramakrishna et al., 2011). In addition, sumoylation showed negative regulation on Nanog expression. Depletion of Sumo1 or its conjugation enzyme Ubc9 significantly increased the expression of Nanog (Wu et al., 2012). Thus, the deletion of PEST motif and depletion of sumoylation showed a high level of Nanog protein stability and expression that could be important findings in iPSC generation. Taken together, the fluctuating expression of Nanog in mouse ESCs (Chambers et al., 2007) might be due to regulation by its PTMs such as phosphorylation and ubiquitination during pluripotency. The conserved domains of human Nanog and the role of PTMs are summarized in Figure 1.
Importance of PTMs of Reprogramming Factors in Tumorigenesis
Several studies have demonstrated that reprogramming transcriptional factors, such as Oct3/4, Sox2, c-Myc, Nanog, and Klf4, plays a crucial role in the malignant development and progress of numerous types of tumors. Prominent expression of reprogramming transcription factors is associated with well-differentiated tumors compared with the undifferentiated tumors (Ben-Porath et al., 2008). There is a close relation between tumor cells and stem cells because several genes associated with ESC maintenance might also contribute for maintaining nondifferentiated state in cancer cells. This fact has been supported by several reports on reprogramming factors showing an unique link with the progression of cancers (Andres et al., 1988; Gidekel et al., 2003; Hochedlinger et al., 2005; Li et al., 2004; Rodriguez-Pinilla et al., 2007; Santagata et al., 2007; Shachaf et al., 2004).
The c-Myc proto-oncogene, together with L-Myc, N-Myc, s-Myc, and B-Myc, belongs to a family of related genes (Henriksson and Luscher, 1996). c-Myc was initially recognized as a cellular homolog of the v-Myc oncogene (Dang, 1999). c-Myc was found to be a target for activation by chromosomal translocation in Burkitt's lymphoma, suggesting its importance in carcinogenesis (Battey et al., 1983). c-Myc, which is responsible for maintaining stem cells, is involved in tumor cell differentiation (Shachaf et al., 2004). The c-Myc gene encodes a short-lived nuclear phosphoprotein consisting of over 430 amino acids. The transactivation domain is found at the amino terminal (∼150 amino acid residues), whereas the DNA-binding domain is found at the carboxyl terminal (90 amino acid residues). Additionally, c-Myc has a dimerization domain allowing it to associate with its obligate partner Max, binds to specific E-box sequences, and is responsible for controlling a set of genes responsible for cell growth and proliferation (Grandori and Eisenman, 1997; Henriksson and Luscher, 1996). The activation or inactivation of the c-Myc gene alone can lead to the formation or regression of liver cancers, indicating that c-Myc is a critical promoter of cell proliferation (Shachaf et al., 2004). Homozygous inactivation of c-Myc in fibroblasts significantly reduces their rate of proliferation by prolonging both the G1 and G2 phases of the cell cycle (Mateyak et al., 1997). Thus, c-Myc exhibits its biological functions by regulating the expression of target genes that are involved in cellular proliferation.
Ser-62 phosphorylation is a critical step for GSK3 phosphorylation at Thr-58, which regulates c-Myc–mediated apoptosis (Lutterbach and Hann, 1994). A mutation at the Thr-58 or Ser-62 phosphorylation sites causes a delay in cell cycle exit, suggesting their roles in cell cycle progression (Kenney et al., 2004). Chang et al. showed that c-MycS62A mutant was weak oncogenically compared to the wild type, resulting in decreased transformation activity (Chang et al., 2000). c-MycS62A was not effective in substituting for small T antigen, along with Ras and telomerase, because the mutant form was not able to cause tumors in immunodeficient mice (Yeh et al., 2004). The cMycS62A mutant was unable to repress thrombospondin-1, an antiangiogenic factor that is necessary for tumor formation, suggesting Ser-62 phosphorylation of c-Myc role in angiogenesis during tumorigenesis (Watnick et al., 2003). A study by another group showed that oxidative stress induces Ser-62 phosphorylation and further activation of the glutathione-mediated survival pathway (Benassi et al., 2006). Thus, Ser-62 phosphorylation is necessary for efficient cell progression and also exhibits resistance to oxidative damage and survival during tumorigenesis (Benassi et al., 2006). However, the phosphorylation at Ser-71 is more significant than that at Ser-62 in regulating the proapoptotic behavior of c-Myc (Noguchi et al., 1999). Thus, S62 phosphorylation on c-Myc was an essential PTM for the tumor development and progression.
Oct3/4 expression is critical in maintaining ESC character and is also capable of inducing tumorigenesis (Hochedlinger et al., 2005). Wild-type expression of Oct3/4 maintains the pluripotent phenotype, whereas two-fold higher than the normal level leads to differentiation of cells (Niwa et al., 2000) and a 1.5-fold higher than the normal level in germ cells leads to gonadal tumors (Looijenga et al., 2003). The ectopic expression of Oct3/4 on somatic tissues results in dysplastic growth in epithelial tissues and increased β-catenin transcriptional activity, leading to tumor growth (Hochedlinger et al., 2005). The expression of Oct3/4 was demonstrated in all human testicular germ cell tumors, and its high expression level is associated with genesis of tumors, whereas Oct3/4 inactivation blocks the signals of malignant components (Gidekel et al., 2003). Oct3/4 is a specific marker for seminoma and embryonal carcinoma (Santagata et al., 2007). In addition, high Oct3/4 mRNA expression level was found in breast cancer cell lines such as MCF7 and MDA-MB-231 (Ling et al., 2012). Thus, the expression level of Oct3/4 plays a critical role in the genesis of various tumors. It has been reported that an auto regulatory mechanism occurs within Oct3/4, which might regulate cell proliferation and differentiation (Kim et al., 2008). Thus, we postulate that the function of PTMs on Oct3/4 plays a critical role in determining the fate of the cell. Ubiquitination and phosphorylation of Oct3/4, which is very essential to regulating Oct3/4 transcriptional activity and its target gene promoters, might also balance the expression level of Oct3/4 to determine pluripotency or induce tumorigenesis.
Other reprogramming transcriptional factors, like Sox2, Nanog, and Klf4, which are key regulators of ESC identity, showed high expression in specific human cancer types (Le Magnen et al., 2012; Li et al., 2004; Liu et al., 2012; Santagata et al., 2007). Sox2 showed higher expression in human gastric carcinomas (Li et al., 2004), basal-like breast carcinomas (Rodriguez-Pinilla et al., 2007), and breast cancer cell lines (Ling et al., 2012) and is a specific marker of embryonal carcinoma (Santagata et al., 2007). Nanog showed high expression in seminomas and embryonal carcinoma (Santagata et al., 2007). Additionally, Nanog is expressed in breast cancer cell lines such as MCF7 and T-47D cells (Ling et al., 2012). Recently, Nanog expression was shown to be critical in human esophageal cancer development (Yang et al., 2012). However, there are no reports on the PTMs of Sox2 and Nanog showing direct association with tumorigenesis. Akt-mediated Sox2 phosphorylation at Thr-118 enhances Sox2 protein stability, and acetylation at K75 regulating protein level might play a major role in maintaining the undifferentiated state in cancer cells. In addition, we postulate that ubiquitination of Sox2 and Nanog, which is responsible for protein turnover, plays a critical role in determining the fate of the cells.
KLF4 along with other reprogramming transcription factors showed high expression in prostate cancer and benign prostatic hyperplasia (Le Magnen et al., 2012). Accumulated evidence showed that KLF4 might function either as an oncogene or a tumor suppressor (Rowland and Peeper, 2006). Increased KLF4 is associated with primary breast ductal carcinoma and oral squamous cell carcinoma (Foster et al., 2000; Foster et al., 2005). In contrast, KLF4 expression was reduced in several cancer types (Rowland and Peeper, 2006; Wei et al., 2005; Zhao et al., 2004). Similarly, overexpression of Klf4 showed inhibition of colonic and gastric tumor growth both in vivo and in vitro (Dang et al., 2003; Wei et al., 2005). Transforming growth factor-β (TGF-β) enhances KLF4 protein turnover, which was confirmed by the inhibitory effect of MG132 on TGF-β–induced KLF4 protein degradation (Hu and Wan, 2011). Cdh1/APC, an E3 ubiquitin ligase, interacts with Klf4 regulating TGF-β–induced KLF4 proteolysis. Thus, the stabilized KLF4 impaired TGF-β–induced transcriptional activation and further antagonized the TGF-β–induced growth inhibition (Hu and Wan, 2011). Additionally, SUMO-1 interacts with KLF4 and sumoylates at K275. The transactivation and antiproliferative activity by KLF4 were directly regulated by SUMO-1 expression (Du et al., 2010). Taken together, the functional roles of KLF4, as both a tumor suppressive and oncogenic gene, might be regulated by several PTMs, depending on the challenges faced to maintain cellular homeostasis. Further studies on the functional mechanisms of PTMs on reprogramming transcriptional factors regulating several signal transductions for tumor development will help us to clearly understand its biological significance during tumorigenesis.
Future Directions for Generating Safe Integration-Free iPSCs
A growing body of evidence supports the view that PTMs on reprogramming transcriptional factors play a major role in regulating transcriptional activity, protein activity, localization, expression, proteolysis, and various biological functions. In the past 6 years, groundbreaking research on the generation of iPSCs has provided several research groups with a unique platform on which to elucidate the mechanisms of cellular reprogramming and the importance of PTMs on reprogramming transcription factors to enhance the stem cell reprogramming efficacy. For example, phosphorylation of Sox2 significantly contributes to the reprogramming of MEFs by improving the efficiency for inducing iPSCs (Jeong et al., 2010), while phosphorylation of Nanog regulates the ESC self-renewal pathway (Moretto-Zita et al., 2010). So far, established methods to generate iPSCs require either genetic materials or mutagenic molecules that are not safe for developing cell therapy for human diseases. Therefore, it is very essential to develop safer methodologies for iPSC generation to have a significant impact on human clinical trials.
In this context, virus-free successful reprogramming has been achieved. A new method of generating human iPSCs by direct delivery of reprogramming recombinant proteins was developed. These reprogramming recombinant proteins are delivered into cells by conjugating them with a short peptide that mediates protein transduction. Zhou et al. conjugated a poly-arginine (11R) transduction domain to the carboxyl terminus of four reprogramming factors, namely Oct3/4, Sox2, Klf4, and c-Myc, along with small chemical molecules treatment (Zhou et al., 2009). These reprogrammed cells were called protein-induced stem cells (piPSCs). In contrast, a system to generate human iPSCs was introduced using direct delivery of reprogramming proteins without the assistance of chemical treatment (Kim et al., 2009). Alternatively, whole cell extracts isolated from ESCs have been used to generate iPSCs (Cho et al., 2010). Protein-based iPSCs were very efficient in generating functional dopamine neurons compared to virus-based iPSCs and could significantly rescue motor deficits in the Parkinson disease model (Rhee et al., 2011). However, the generation efficiency of piPSCs is slow, with approximately double the time taken for viral transduction–mediated iPSCs, and inefficient.
A recent trend is to generate iPSCs either by activation of reprogramming transcription factors with the treatment of mRNA-encoding reprogramming factors (Plews et al., 2010) or by a novel synthetic mRNA-based approach to obtain miRNA derived iPSCs (Heinrich and Dimmeler, 2012). Recently, integration-free human iPSCs were generated using episomal plasmid vectors, including reprogramming transcriptional factors (Okita et al., 2011).
Recently, Deng's group developed a novel chemical induction method for generating iPSCs, which is promising for clinical use. Initially, iPSCs were generated using single-gene Oct3/4 along with a combination of small molecules termed as VC6T [VPA,CHIR99021 (CHIR), 616452, and tranylcypromine] (Li et al., 2011). Later, the same group screened for nearly 10,000 small molecules that could substitute Oct3/4 as supplements to the VC6T chemical combination to obtain transgene-free iPSCs, called CiPSCs. First, VC6T plus forskolin (VC6TF) was used on the Oct4 promoter-driven green fluorescent protein (GFP) expression system, which turned out to be incomplete reprogramming (Li et al., 2011). Later, several cyclic adenosine monophosphate (cAMP) agonists, such as forskolin, prostaglandin E2, and rolipram, and epigenetic modulators, such as 3-deazaneplanocin A (DZNep), 5-azacytidine, sodium butyrate, and RG108, were analyzed on a doxycycline-inducible Oct3/4 expression system. Among them, DZNep, a compound known to catalyze late reprogramming, showed a significant improvement in generating iPSCs (Hou et al., 2013). At 16 days after treatment with VC6TF, DZNep was added, and 28 days later cells were fed with 2i-medium for dual inhibition (2i) of GSK-3 and mitogen-activated protein kinase (MAPK) signaling, resulting in completely reprogrammed ESCs. Furthermore, addition of the synthetic retinoic acid receptor ligand TTNPB boosted the reprogramming efficiency up to 0.2%, which was comparable to those obtained from standard iPSC production methods (Hou et al., 2013). Thus, chemically induced (Ci) PSCs are safe and feasible considering that the chimeric mice generated from CiPSCs were 100% viable and healthy. However, translation of this technique to human somatic cells requires further improvements and screening of additional small molecules to meet the needs of regenerative medicine. Taken together, chemicals used to generate CiPSCs are safe and do not risk causing mutations. The small molecules can readily penetrate cell membranes, so they can be washed away after they induce cellular reprogramming. However, to develop a safe method of generating patient-specific stem cells based on mRNA, protein, or chemical iPSC technology, one should thoroughly understand the role of PTMs on defined reprogramming proteins and how their biological functions are regulated, as summarized in this review.
Areas of future investigations include identification of PTM sites that increase the stability of these reprogramming proteins. Replacing critical amino acid residues of reprogramming proteins responsible for protein degradation and transactivation suppression might improve the reprogramming efficiency without affecting its pluripotency function. It is known that these reprogramming proteins undergo ubiquitination, which is a crucial regulatory mechanism involved in several fundamental biological functions, including cell differentiation. In the cascade of enzymes involved in proteasome degradation pathway, E3 ligase plays a critical role in transferring ubiquitin from an E2 enzyme to the target protein, resulting in protein degradation. Therefore, identification and blocking E3 ligases specific for reprogramming transcriptional factors increases the protein expression level posttranscriptionally in stem cells, finally leading to rapid and efficient generation of patient-derived human iPSCs.
Alternatively, identification of specific deubiquitinating enzymes (DUBs) for reprogramming proteins, a class of proteins that are involved in the removal of ubiquitin molecules from the protein that are targeted for protein degradation, represents a potentially promising method for enriching reprogramming proteins during iPSC generation. Herpesvirus-associated ubiquitin-specific protease (HAUSP) promotes maintenance of neural stem/progenitor cells by deubiquitinating repressor element 1–silencing transcription factor (REST), which is an essential transcriptional repressor of neuronal differentiation (Huang et al., 2011). The protein stability for each reprogramming transcriptional factor might be regulated by more than one specific DUB and E3 ligase for cell fate determination of stem cells (Fig. 2). Thus, identification and modifications of such stem cell regulators might provide a novel method to maintain pluripotency of stem cells. Along these avenues of investigation, improved and stable reprogramming proteins are obtained that can facilitate generation of efficient and safe patient-specific human iPSCs.

A model showing the regulation of E3 ligases and deubiquitinating enzymes (DUBs) on reprogramming transcriptional factors for cell fate determination of stem cells.
Taken together, genetic manipulations to regulate the PTMs of these reprogramming proteins represent potential alternatives for improvising the stability and efficiency of these reprogramming proteins. This might lead to the development of a promising human iPSC technology that is free from cancer development for drug discovery, disease modeling, and future clinical trials.
Footnotes
Acknowledgments
We would like to thank Drs. Bharathi Suresh and Kimberly Kim for their critical comments on the manuscript. We regret that we were unable to cite all relevant studies because of space constraints. This work was supported by Basic Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2011-0015312).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.