
Abstract
Abstract
Four normal-karyotype human embryonic stem cell (hESC) lines were generated using the same protocol and maintained under identical conditions. Despite these precautions, gene expression patterns were found to be dissimilar among the four lines. The observed differences were typical of each cell line, correlated with their distinct propensity to exit stemness, created heterogeneity among the cells during cell line maintenance, and correlated with their altered capacity as a source of differentiated cells. The capacity of some cell lines to give rise to more, and more mature, neurons within comparable time frames of directed differentiation reflected the distinct proportions of cells already predifferentiated at the onset. These findings demonstrate that the subsequent stages of neural differentiation were altered both in a quantitative and timely fashion. As a consequence, cell lines with apparent better and quicker ability to produce neurons were actually the less capable of reproducing proper differentiation. Previous data suggested that cell lines able to generate more neurons faster would be more suitable to clinical application. Our analysis of the differentiation process strongly suggests the opposite. The spontaneous tendency to predifferentiate of any particular hESC line should be known because it clearly impacts further experimental results.
Introduction
T
The causes of the variability of gene expression patterns among hESC lines are unknown. The different genetic background of the human embryos used to derive the lines or differences in the culture conditions used during derivation and/or expansion of the cell lines rank among possible causes (Skottman et al., 2006; Tavakoli et al., 2009). These differences are likely to impact on any experimental outcome, while the practical consequences still need to be identified. The capacity of some hESC lines to give rise to more neurons after an identical period of directed differentiation has been documented (Lappalainen et al., 2010; Tavakoli et al., 2009), but the degree of different hESC lines all derived under very similar conditions to enter the neural differentiation process in a synchronized way, a prerequisite to obtain a high rate of defined neuron-subtypes, has never been analyzed in detail.
Four new hESC lines were generated in our institution using the same protocol. This gave us the opportunity to address the question of inherent versus derivation-dependent or culture-dependent differences in gene expression and differentiation behavior of hESC lines. Although the derivation and maintenance of the hESC lines were carried out under very similar conditions, inherent differences in their gene expression patterns became soon evident. These differences correlated with their distinct tendency to predifferentiate spontaneously during stem cell maintenance. We describe several aspects of their differentiating behavior, found to be altered as a result of an initial greater propensity of some cells to predifferentiate. We discuss those observations in terms of care in the choice of cell lines adapted to specific diagnostic or therapeutic purposes.
Materials and Methods
Regulatory aspects and laboratory environment
All procedures related to the donation of surplus embryos deriving from assisted reproduction technology (ART) and their use for research purposes were in accordance with the Swiss law on ART and the Swiss law on human ESC research. This project was approved both by the local ethics committee (EKBB) and by the Swiss Federal Office of Public Health (BAG) (see Supplementary Data available at www.liebertpub.com/cell/). All experiments were carried out in a laboratory equipped with grade A, particle–free technology. This laboratory is separated from, but in close proximity to, the ART unit. All procedures and working conditions in the embryology laboratory of the ART unit were modified to xeno-free conditions, including full traceability of the components of all processes.
hESC derivation and culture
After fertilization, embryos were cultured at 37°C in a humidified gas mixture containing 6% CO2 and 6% O2 for 3 days in K-SICO (Cook Medical, www.cookmedical.com), without medium changes in 25-μ L droplets under oil (K-SIMO; Cook Medical). On day 3, the medium was changed to K-SIBO (Cook Medical). On day 6 or 7, the donated blastocysts were mechanically disrupted into two pieces. The pieces with the ICM were further cultured in hESC medium on a human feeder layer [35 Gy γ-irradiated foreskin fibroblasts; American Type Culture Collection (ATCC CRL2429)]. After 6–8 days of culture, the outgrowth was mechanically split into six to 10 pieces and again placed onto a new feeder layer. After further 6–8 days, the first hESC colonies started to appear. For stem cell culture, knockout DMEM-KO culture medium (Invitrogen, catalog no. 10829-018) was supplemented with 20% KSR (Invitrogen, catalog no.10828-028), 2 mM L-GlutaMax (Invitrogen, catalog no. 35050-038), 0.05 mM β-mercaptoethanol (Invitrogen, catalog no. 31350-010), 0.1 mM nonessential amino acid (Invitrogen, catalog no. 11140-035), and 8 ng/mL basic fibroblast growth factor (bFGF; R&D Systems, catalog no. 234-FSE-025/CF) in the presence of penicillin (50 U/mL) and streptomycin (50 μg/mL; Invitrogen catalog no. 15140-122) and is denominated henceforth hESC-M.
Characterization of hESC lines with the alkaline phosphatase assay and with immunohistochemistry
Alkaline phosphatase (AP) staining and immunofluorescence were performed according to the recommendations provided by the manufacturer (Millipore, catalog no. SCR001). For AP-staining, ESCs were cultured for 4–5 days, fixed, and incubated in the staining solution Fast Red Violet for 15 min. For immunofluorescence staining, cells were incubated with primary antibodies against the stemness markers Oct-4, SSEA-4, TRA-1-60, and TRA-1-81 at 1:200 (Millipore, catalog no. AB3209). Secondary antibodies fluorescein isothiocyanate (FITC)-labeled goat anti-mouse immunoglobulin M (IgM)/IgG (Sigma, catalog no. NF-1010) or tetramethylrhodamine (TRITC)-labeled donkey anti-rabbit IgG (Sigma T-6778) were used at 1:200 for 30–60 min at room temperature.
In vivo differentiation analysis
Eight- to 10-week-old immune-incompetent nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (Charles River Laboratories, Germany) were used to demonstrate the in vivo differentiation potential of the newly derived hESC lines. Housing and experimental protocols were approved by the Basler Cantonal Veterinary Department and were in accordance with Swiss Animal Protection Law. About 100–200 cells from 60 colonies of the CHES2, CHES3, or CHES5 line were mixed with Matrigel (BD Biosciences, catalog no. 354277) and injected subcutaneously into two NOD/SCID mice. Two months later, the mice were sacrificed, the tumors removed for Hematoxylin & Eosin (H&E) staining and histological evaluation with a conventional light microscope (Dialux 20; Leitz, Zeiss Axio Imager A.1). Immunohistochemistry was performed on a Ventana Immunostainer using Cytokeratin (CK22, Biomeda,1:500), neuron-specific enolase (NSE), desmin (DES), vimentin (VIM; Ventana/ Roche), and smooth muscle actin (SMA; DAKO), antibodies prediluted by the manufacturer.
Differentiation in vitro
For random differentiation into the three germ layers, hESC colonies were mechanically scraped off the feeder layer. To form embryoid bodies (EBs), two to three pieces, each consisting of around 50–100 cells, were placed into 50-μL droplets of hESC medium without bFGF. Some EBs were analyzed after 4 days by quantitative PCR (qPCR). For further differentiation, the EBs were plated onto a gelatinized dish and cultured for 8–14 days in the hESC medium in which FCS was substituted for KSR.
For directed neural differentiation, the colonies of each hESC line were removed from the feeders, dissociated into small clumps, and expanded in suspension in low-attachment-well plates (HydroCell™, Nunc, catalog no. 2014-05) (Steiner et al., 2010). The cells were amplified in hESC-PRO medium corresponding to neurobasal medium (Invitrogen, catalog no. 21103049) supplemented with 14% knockout DMEM-KO culture medium (Invitrogen, catalog no.10829-018), 1×nutridoma-CS (Roche, catalog no. 11363743001), 2 mM L-GlutaMax (Invitrogen, catalog no. 35050-038), penicillin (50 U/mL), and streptomycin (50 μg/mL; Invitrogen, catalog no. 15140-122), 0.1 mM nonessential amino acids (Invitrogen, catalog no. 11140-035), 25 ng/mL recombinant human Activin A (Prospec CYT-569), 0.5 μg/mL human laminin (Sigma, catalog no. L4544), and 20 ng/mL recombinant human bFGF (R&D Systems, catalog no. 234-FSE-025/CF). Clumps were partially dissociated once a week by gentle shaking and cultured in the presence of 10 μM ROCK inhibitor (Sigma, catalog no. Y0503) for 1 day. For one experiment, hESC-PRO was substituted with TeSR1 (StemCell Technologies, catalog no. 05850). To start neuroectodermal induction, the clusters were gently dissociated and plated on 5 μg/mlL (collagen IV, Sigma, catalog no. C5533) at a density of 90,000 cells/cm2 and cultured in neural differentiation medium consisting of Dulbecco's modified Eagle medium (DMEM)/F12 (Invitrogen, catalog no. 31331-028), 1×N-2 Supplement (Invitrogen, catalog no. 17502-048), 0.1 mM nonessential amino acids, penicillin (50 U/mL), and streptomycin (50 μg/mL; Invitrogen, catalog no. 15140-122), and 600 ng/mlL recombinant human noggin (Prospec CYT-475) for 7 days. The cells were then further cultured for an additional 2 weeks in the same medium but in the absence of noggin. Immunofluorescence was performed using mouse anti-βIII tubulin (1/200; Sigma, catalog no. T5076) or rabbit anti-βIII tubulin (1/200; Covance, catalog no. PRB-435P), rabbit anti-MAP2 (1/50; Sigma, catalog no. HPA 012828), and mouse anti-Nestin (1/50; R&D Systems, catalog no. MAB1259). Secondary antibodies Alexa Fluor 488 or 546 conjugated with either anti-mouse or anti-rabbit IgGs were used at 1/1000. The nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI; Sigma, catalog no. 32670).
Quantitative PCR
Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel, catalog no. 740955) according to the manufacturer's instructions. For each sample, 300 ng of total RNA was reverse-transcribed into single-stranded cDNA by the SuperScript®II Reverse Transcriptase (Invitrogen, catalog no. 18064.014) in presence of 0.5 μM of deoxynucleotide triphosphates (dNTPs; Invitrogen, catalog no. 10297-018) and a mix of 5 ng/μL random hexamer primers (Invitrogen, catalog no. 48190-011).
One-tenth of each cDNA was used for each PCR reaction, performed following the guidelines given by the manufacturer (SABiosciences; Embryonic Stem Cells PCR Array, catalog no. PAHS-081) by using Power SYBR® Green PCR Master Mix (Applied Biosystems, catalog no. 4367659) in an ABI 7500 Fast Real-Time PCR System (Applied Biosystems). For each sample, two qPCR measurements were performed, and the average was used to calculate the change fold. The qPCR primers used were synthesized by Microsynth (Balgach, Switzerland) and are described with their used annealing temperature in Figure S4 (Supplementary Data are available at www.liebertpub.com/cell/).
Results
Establishment of four new hESC cell lines
From August, 2008, to August, 2010, a total of ∼800 infertile couples were treated with ART. During this time interval, 15 blastocysts were donated for hESC derivation, but only seven were used with our standard derivation protocol (see Materials and Methods). All donated embryos arose from pronucleate oocytes that were cultured after intracytoplasmic sperm injection and developed up to the blastocyst stage. Four hESC lines were generated from those seven embryos following a fixed and established protocol (Inzunza et al., 2005).
Between days 6 and 8 after fertilization, the hatched blastocysts were cultured on a human foreskin fibroblast feeder layer (CRL2429). Three seeded blastocysts produced no outgrowth (Table 1). The growing cell mass of the remaining four blastocysts was cut mechanically into six to 12 pieces and further cultivated on a feeder layer for another 6 days. When they developed ESC-like colonies, they were separated and grown over prolonged time periods. All four lines, CHES2, CHES3, CHES5, and CHES6, have been fully characterized and registered, and their characteristics have been published in the European Human Embryonic Stem Cell Registry (hESCreg).
Grading of blastocyst according to Gardner et al. (2000).
Cell cluster forming after embryo plating on feeder.
hESC, human embryonic stem cell; ICM, inner cell mass; ZP, zona pellucida.
Cells were karyotyped using standard GTG banding metaphase spreads, and normal chromosome complements were found in all four hESC lines (Fig. 1A–D). For CHES2, a comparative genomic hybridization was performed at passage 22 to detect rearrangements or multicopy amplifications, but no aberrations were detected. All cell lines (CHES2 at passage 70, CHES3 at passage 40, CHES5 at passage 40, and CHES6 at passage 48) were characterized with immunohistochemistry, and the expression of a number of typical stemness markers, such as AP, TRA60, TRA81, SSEA-4, and OCT-4, was confirmed (Fig. 1E–H). These hESC lines were capable of undergoing in vitro differentiation to EBs containing cells belonging to the three germ layers (see Fig. S1). These EBs can then be further differentiated into cardiomyocytes and neurons (data not shown). For full characterization of these cell lines, teratomas were formed in vivo showing derivative tissues of all three germ layers as well (Fig. 1I–M).
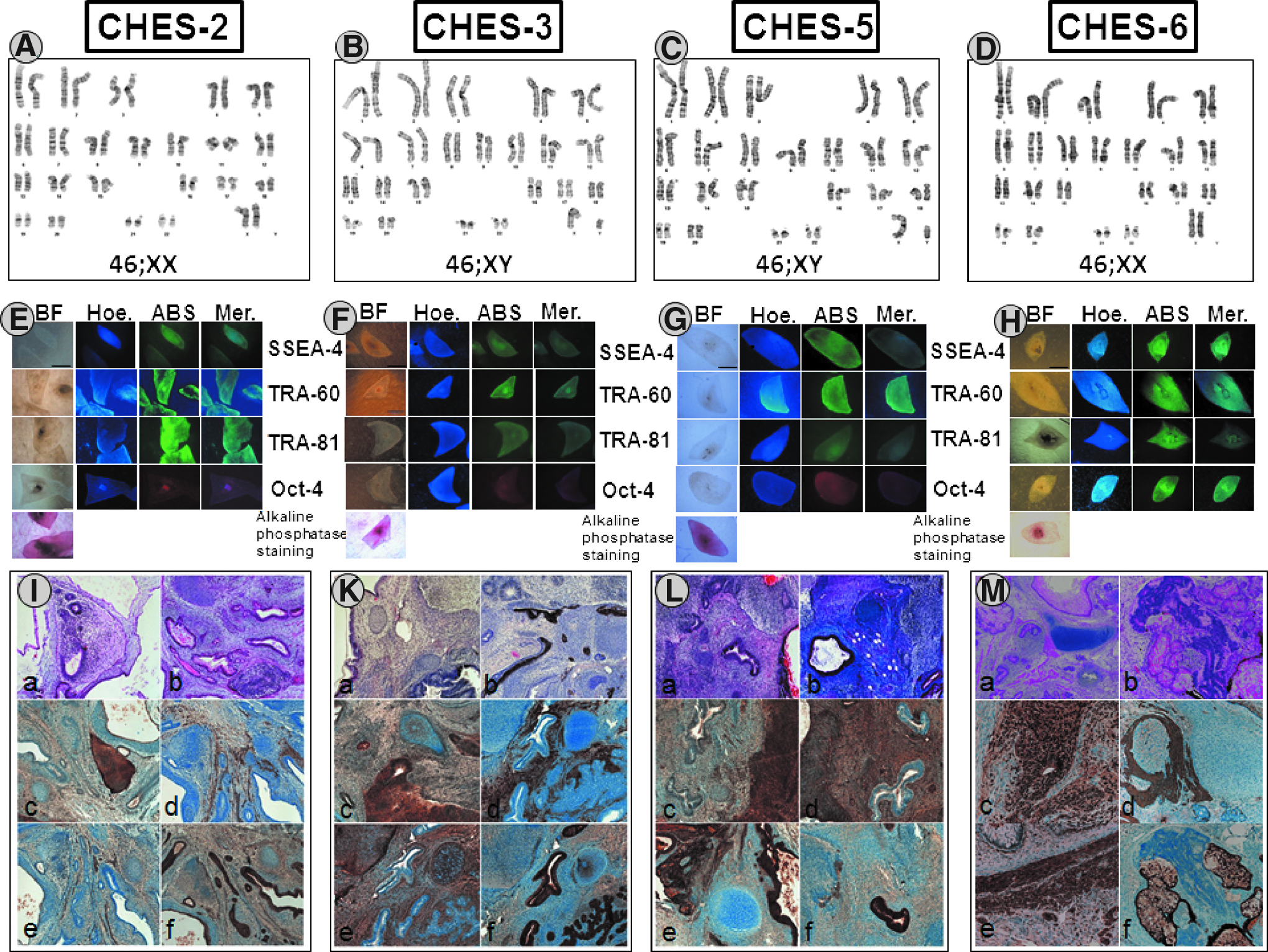
Characterization of new established lines CHES2, CHES3, and CHES5 by karyotyping, immunohistochemistry of the hESCs and histological teratoma analysis (
Comparison of the gene expression patterns of the newly derived hESC lines
Three hESC lines were then characterized by qPCR array, including 44 ESC-specific genes and 40 selected ESC differentiation genes (see Fig. S2). CHES3 exhibited a gene expression profile similar to the commonly used reference line HS401 (Table 2) (Martin-Ibanez et al., 2008). For none of the genes examined in both cell lines was a more than three-fold difference in the expression levels detected. In contrast, when compared to HS401, four genes were differentially expressed in CHES2 and eight in CHES5. Some are stem cell–specific genes and were differentially expressed at either a higher or lower level: GBX2 (+5.9) in CHES2, GAL (−3.9), GBX2 (+3.4), LEFTY1 (−5.9), NODAL (−4.9), and NOG (+3.5) in CHES5. Furthermore, those two cell lines expressed higher levels of some differentiation genes—neuro-ectodermal markers NEUROD1 (+3.9) and PAX6 (+13.9) as well as the endodermal marker SERPINA1A (+4.1) were overrepresented in CHES2, whereas PAX6 (+4.7) and the trophoblastic marker CDX2 (+3.3) were more expressed in CHES5. In addition, the germ cell marker SYCP3 (−3.6) appeared underexpressed in the latter.
Only genes with three fold regulation are listed.
Fold regulation of new hESC line compared to HS401.
The expression levels of these genes in the hESC lines were also compared with those of neonatal foreskin fibroblasts to account for any potential contamination by the feeder cells (see Fig. S3). When compared to fibroblasts, a number of pluripotency markers were highly overexpressed in all hESC lines (GAL 67–260 times, LEFTY1 110–652 times, NODAL 38–189 times, GBX2 4.2–25 times), whereas Nog was overexpressed more than three times only in CHES2 and CHES5. However, other stem cell markers did not appear to be overrepresented because they were already expressed at similar (LIFR, TERT, NANOG) or even higher levels (Il6ST, IFITM2, FGF5, and COMMD3) in the foreskin fibroblasts (see Fig. S4c and S4d, respectively).
Some genes classified as differentiation markers (FN1, LAMA1, LAMB1, LAMC1, and DES) were expressed at similar levels in both stem cells and fibroblasts. For that panel of genes, the relative expression profile was not significantly different among the various hESC lines (see Fig. S4b). However, other differentiation markers were clearly overexpressed in hESCs when compared to fibroblasts. The neural marker PAX6 (7.9–110 times), the trophoblast marker CDX2 (3.7–5.3 times), and the visceral endoderm marker SERPINA1 (3.0–8.0 times), as previously found, but also the visceral endodermal marker α-fetoprotein (AFP; 14.9–28.9 times), the trophoblast marker EOMES (45.7–186.8 times), the extraembryonic endoderm FOXA2 (22.8–78.0 times), and the mesoderm marker T-Brachyury (Bra; 19.4–59.4 times) were overexpressed in hESC lines. Those correspond mostly to genes not expressed in fibroblasts (Fig. 2). In the panel of genes used here, each cell line exhibited a different expression profile, which was not influenced by the amount of feeder cell transcripts in the samples (see Fig. S3). Furthermore, the similarities of the expression profiles of HS401 and CHES3 and those of CHES2 and CHES5 were striking.
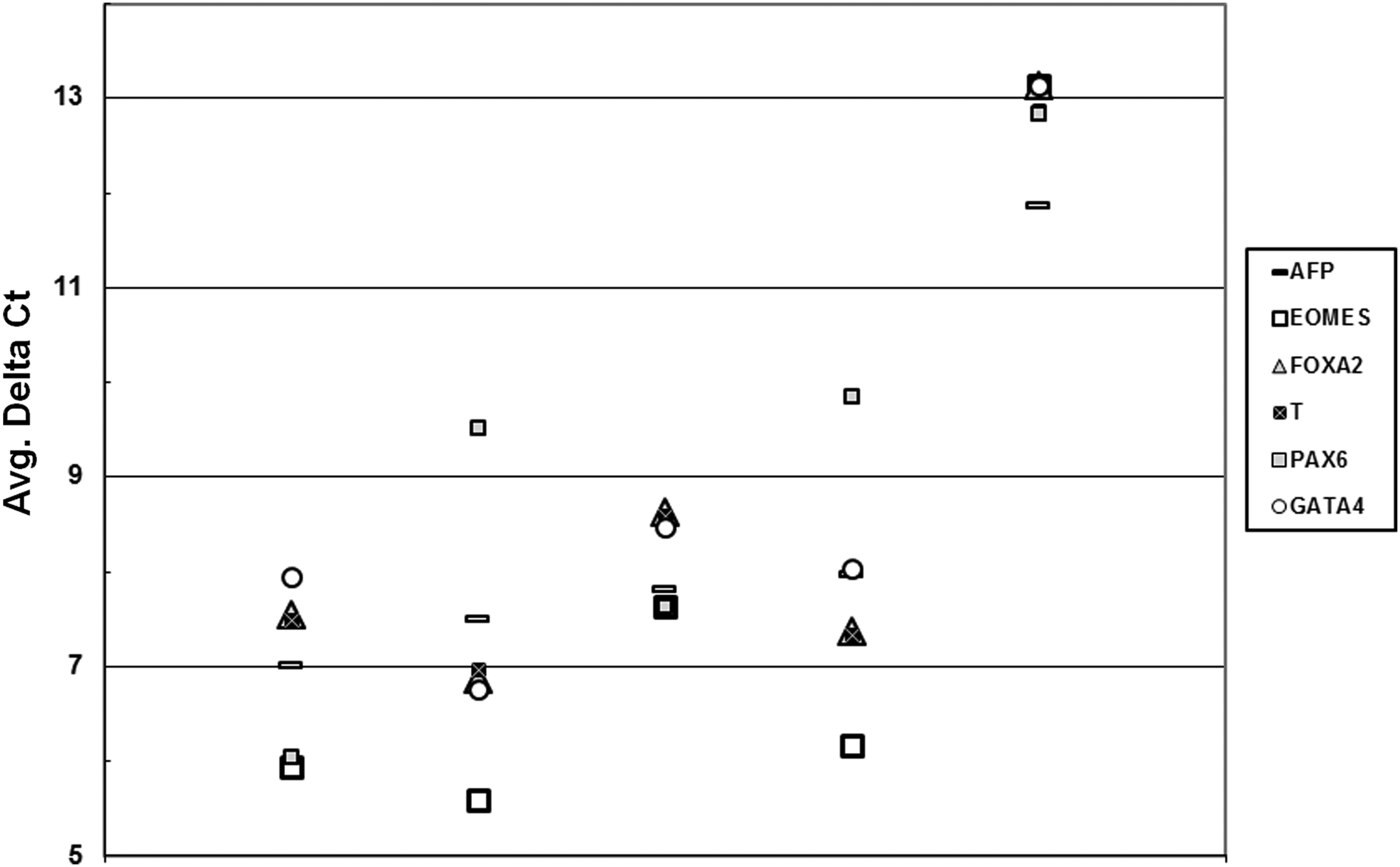
qPCR data generated from qPCR array. Average delta Ct values of CHES2, CHES3, CHES5, HS401, and CRL2429 fibroblasts are shown. Differentiation markers overexpressed in the hESC lines are compared to CRL2429.
Spontaneous differentiation propensity and directed neural differentiation capabilities of the four hESC lines
To test the biological significance of the observed differences in gene expression among the various newly derived hESC lines, we compared their capacity for neural differentiation under identical conditions. As shown in Figure 3A and in accordance with the qPCR array, their different ability to maintain stemness when cultured on fibroblast feeders gave rise essentially to the expression of neuro-ectodermal markers (i.e., Pax6 in the experiment), whereas markers of other layers were detectable by qRT-PCR at a much lower level (i.e., AFP as a marker for endoderm, T-Brachyury for mesoderm, and keratin-5 for nonneural ectoderm).
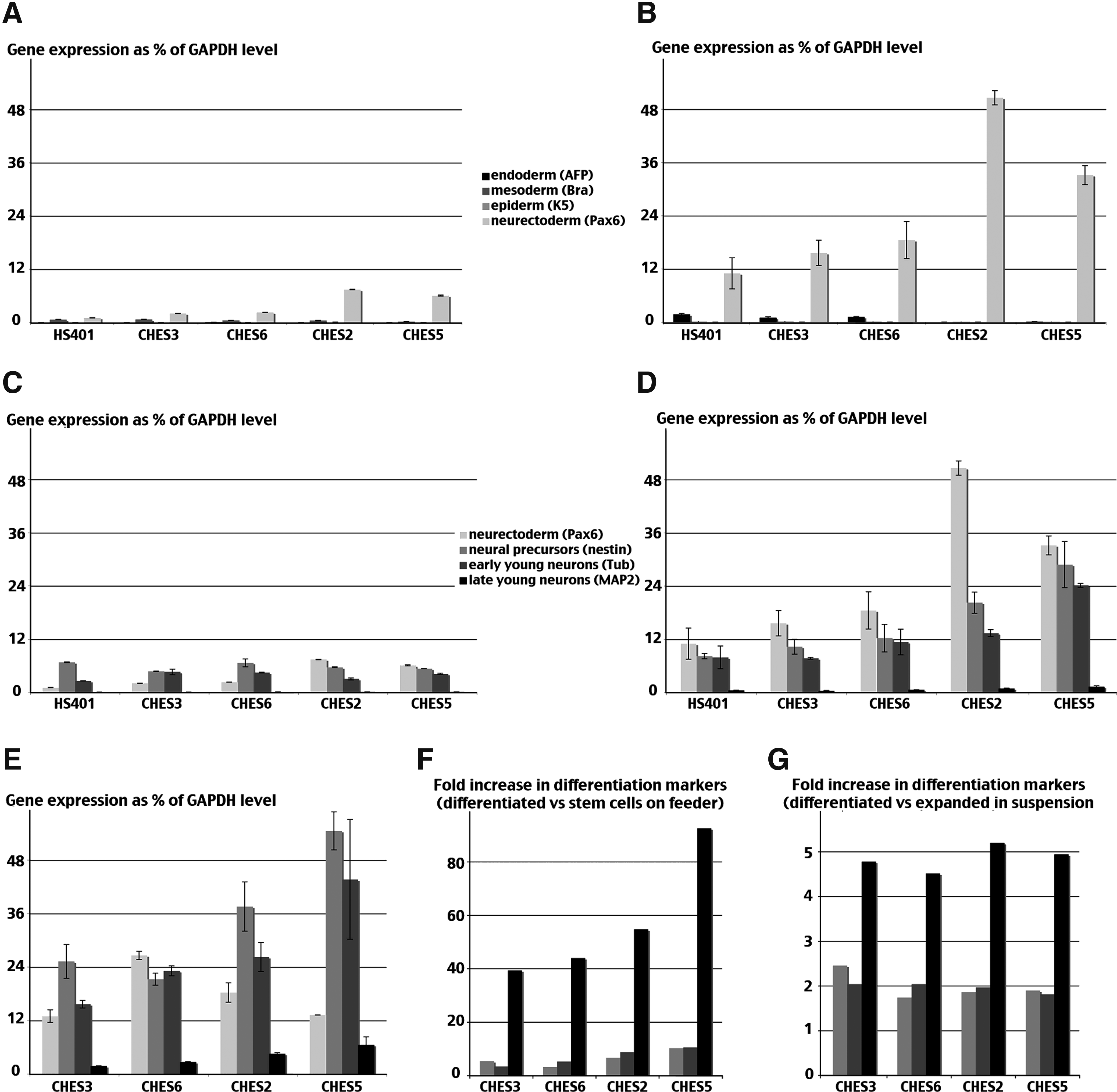
Furthermore, large-scale expansion in a defined microcarrier-free suspension culture system (Steiner et al., 2010) appeared even more permissive because spontaneous neuroectodermal commitment increased while retaining the line-specific differences already observed (Fig. 3B). CHES3 and CHES6 appeared more prone to remain undifferentiated, whereas the CHES2 and CHES5 cell lines had a greater tendency toward spontaneous commitment. In addition, low levels of neural differentiation were also observed in both culture conditions as the marker of neural precursor nestin, and the marker of early neuronal development βIII-tubulin was detected as well (Fig. 3C and D). That predifferentiation tendency appeared similarly constrained for both cell lines when cultured on feeders (Fig. 3C), whereas in the more permissive suspension culture, predifferentiation was more pronounced, especially in CHES2 and CHES5 cell lines, which expressed low but, when compared with the other two lines, significantly higher levels of MAP2, a marker of more advanced neurons (Fig. 3D).
When plated on collagen and cultured in neural differentiation medium, all cell lines displayed a similar pattern of morphological changes typical of neuronal differentiation (not shown). To quantify neuronal differentiation, we measured the relative levels of differentiation markers after 3 weeks in culture. As shown in Figure 3E, markers of both neural precursors and young neurons increased in all cell lines. When compared to the essentially undifferentiated initial cell populations present in cultures on feeder, the expression level of the neural precursor Nestin was five to 10 times higher, and the expression level of the neuronal marker MAP2 was increased 40–90 times. However, CHES5 achieved a level of differentiation twice that of CHES3 and CHES6, whereas CHES2 exhibited an intermediate profile (Fig. 3F). In contrast, when the expression levels of the various differentiation markers at the end of the experiment were compared with their levels immediately before plating, the rate of increase in differentiation markers expression was indistinguishable among the four cell lines (Fig. 3G).
We confirmed by immunocytochemistry that the differences in global expression of differentiation markers indeed reflect distinct capacities of different hESC lines to give rise to neurons during a given time period of directed differentiation. For this purpose we analyzed the percentage of cells expressing those markers in two cell lines (CHES2 and CHES6) with distinct gene expression profiles (Fig. 4). After 8 days in differentiating culture conditions, almost 100% of cells in both lines were actually engaged in the neural lineage, either as neural precursors expressing nestin or as young neurons expressing βIII-tubulin (Fig. 4A). However, CHES6 cells were mostly typical bipolar nestin-positive neural precursors, only around a quarter of the culture being composed of young neurons (Fig. 4, A and B). In contrast, nearly twice as many of the CHES2 cells differentiated to neurons (Fig. 4A), and cells expressing nestin exhibited a more advanced morphology resulting in cells displaying an ambiguous morphological status, halfway between still true precursors and very young neurons but not yet expressing βIII-tubulin (Fig. 4B). Although the number of neurons increased slightly in both cultures during the two subsequent weeks of differentiation, the difference between the two cell lines was only marginally reduced (Fig. 4C). As neurons maturated and progressively co-expressed βIII-tubulin and MAP2, differences in maturation remained clearly visible between both cell lines. After 3 weeks of differentiation, most CHES2 neurons were expressing MAP2, whereas a significant number of CHES6 neurons were still only expressing βIII-tubulin (Fig. 4C and D). These differences in the advancement of neural differentiation among both cell lines are also visible in their morphology: βIII-tubulin and MAP2 coexpressing neurons of CHES2 exhibit more developed extruding processes, which even started to form networks (Fig. 4D). Thus, the higher level of differentiation exhibited by hESC lines with a higher propensity to predifferentiate spontaneously implied both the generation of more neurons and the formation of more mature neurons during the same time interval.
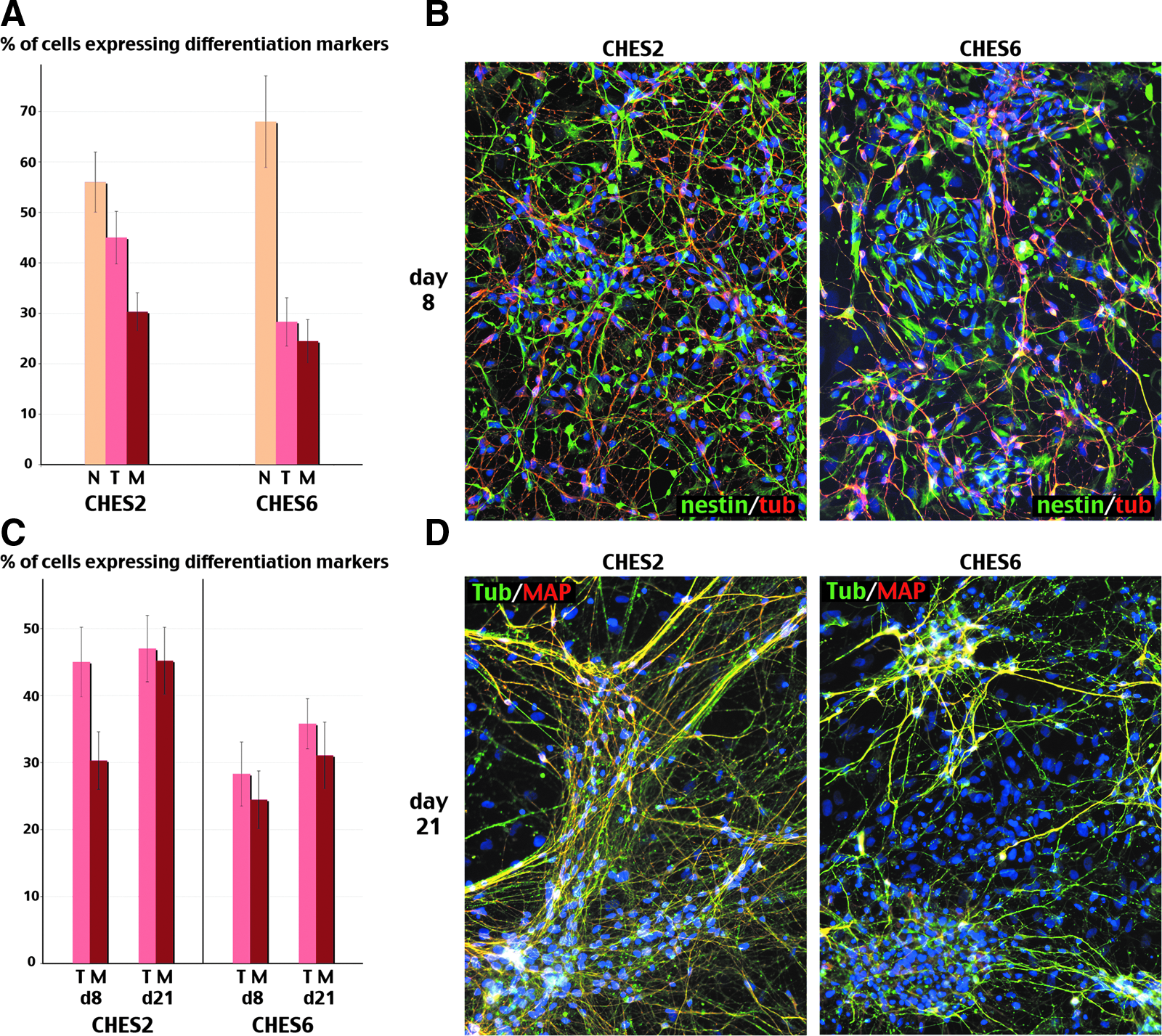
The number of cells expressing neural differentiation markers was determined after 8 (
Consequences of spontaneous predifferentiation for further behavior of neural cell populations during the differentiation process
This property of some hESC lines to predifferentiate was also responsible for an asynchrony in the differentiating cell populations, which rendered more difficult the distinction between the successive phases of the neuronal differentiation process. As clearly visible for CHES6 in Figure 5, in which predifferentiation of the starting population was moderate, those phases could be visualized by following the stepwise evolution of expression of selected markers during differentiation. A transient upregulation of Pax6 expression corresponding to the commitment of stem cells into neurectoderm was followed by a wave of Nestin expression as neuroectodermal cells transformed into neural precursors. Those precursors then gave rise to neurons expressing first βIII-tubulin, then MAP2. In the absence of mitogenic factors needed to sustain the proliferation of precursor cells, such as bFGF, the level of Nestin expression decreased proportionately with the increase in the expression of neuronal markers, indicating the progressive replacement of proliferating progenitors by maturating neurons. In contrast, in cell lines such as CHES5, which was characterized by a more heterogeneous cell population at the onset, the expression profile of each marker was a mix of several overlapping waves (Fig. 5). Nestin expression increased first in already committed Pax6-positive cells and only secondarily in cells newly committed from stem cells. It then correlated first with a decrease in Pax6 expression and persisted while the secondary wave of neurectoderm formation became visible. This gave rise to the paradoxical impression that the order of the various differentiation waves was inverted in CHES5 as compared to CHES6.
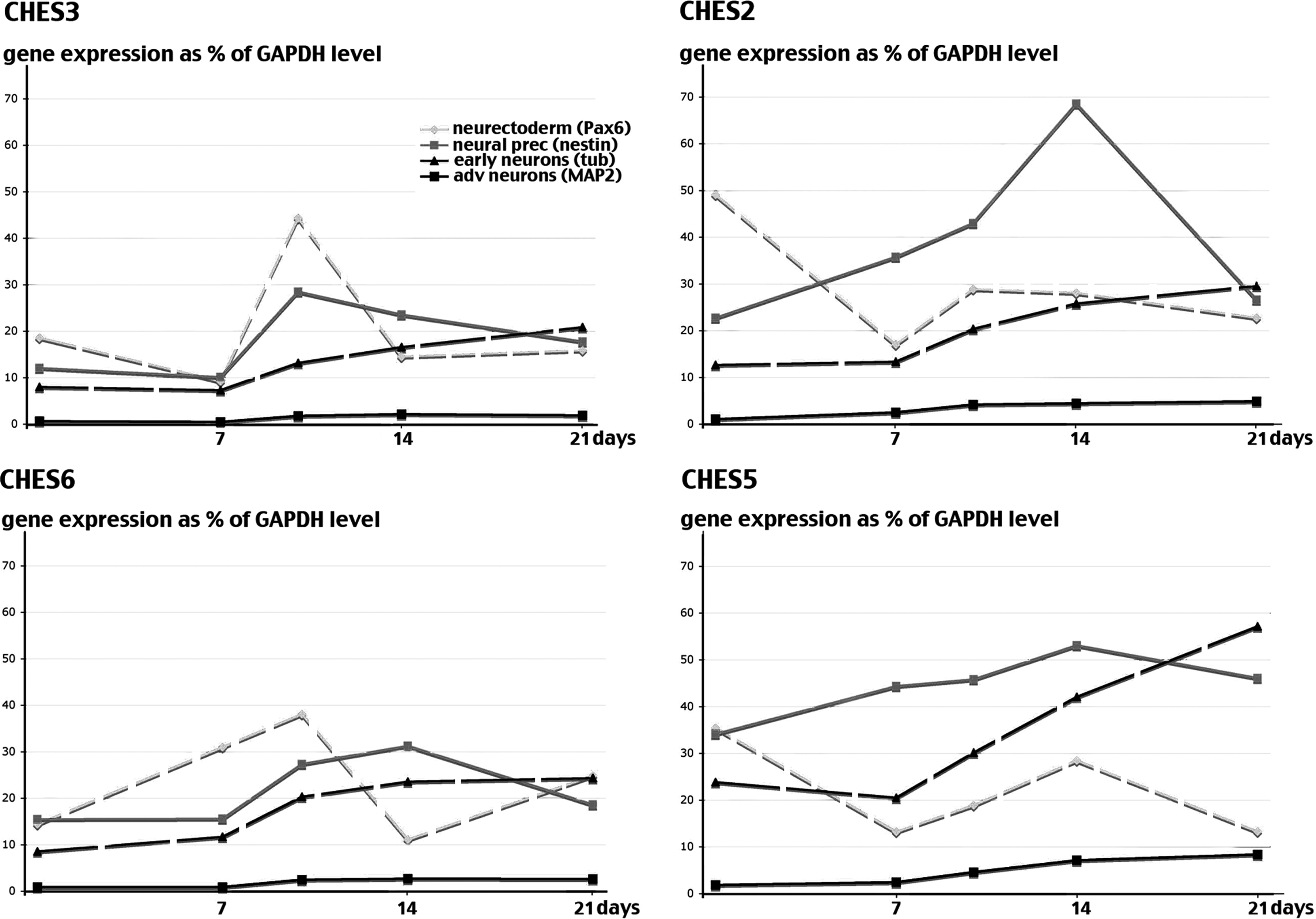
Expression time-course of neural differentiation markers in differentiating CHES2, CHES3, CHES5, and CHES6 cell lines. Expression of neural genes Pax6, nestin, β-tubulin III (Tub), and MAP2 in differentiating CHES2, CHES3, CHES5, and CHES6 lines was quantified 7, 10, 14, and 21 days after initiation of directed neural differentiation (as in Fig. 4).
Variable levels of predifferentiation among different hESC lines were also associated with distinct abilities to form rosettes, the characteristic structures that form in vitro as neural precursors, which acquire the contractile epithelial properties needed to form the neural tube. After 1 week of differentiation, the number of rosettes was similar in all cell lines (Fig. 6A). However, as expected for more advanced cultures, rosette-forming cells in CHES2 and CHES5 expressed higher levels of Sox1, a marker of late mature rosettes in the human (Fig. 6B). After 10 days, the situation had changed radically (Fig. 6C). While the number of rosettes had increased in CHES3 and CHES6, CHES2 and CHES5 had barely progressed to form new rosettes. At that time, they were mostly mature rosettes expressing Sox1 in culture of all cell lines (Fig. 6D). Interestingly, the ability to increase the number of rosettes during that 3-day interval correlated with the ability of differentiating CHES3 and CHES6 cell populations to produce a sharp peak of Sox1 expression, reflecting synchronous differentiation, whereas stagnation of the number of rosettes in CHES2 and CHES5 was associated with rather flat kinetics of Sox1 expression (Fig. 6E).
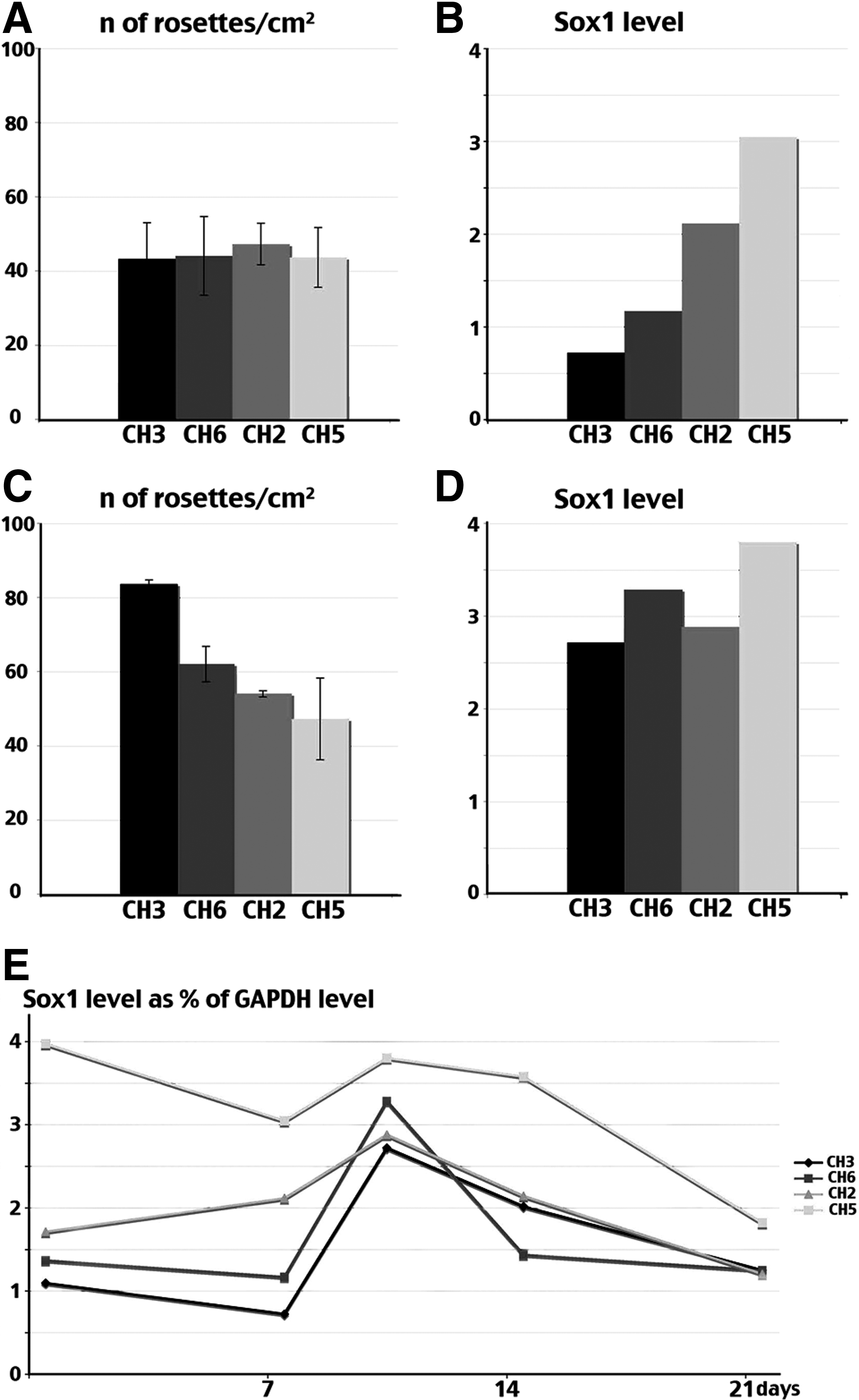
Formation of neuroepithelial tube-like structures (rosettes) during differentiation of CHES2, CHES3, CHES5, and CHES6 cell lines. The number of rosettes formed in differentiating cultures was counted 7 days (
Discussion
Four new hESC lines were derived under very similar, if not identical, conditions. No isolation of the ICM was performed, and there was almost no need for manipulation of the embryo, thereby reducing the mechanical stress. The characterization of the hESC lines by comparative genomic hybridization (CGH) array and immunohistochemistry of both in vitro and in vivo differentiated hESCs did not indicate any abnormalities in the genomic integrity of the cells nor in their stemness properties. Notwithstanding these measures, our data also clearly demonstrated a number of differences in the gene expression patterns among all four hESC lines and the important consequences for their behavior during directed differentiation.
The origin of the variability among hESC lines remains unclear. As with lines derived in different laboratories, these differences could be explained by the distinct embryo culture conditions (day 0–6) used in ART laboratories, similar to what is known in the mouse (Fernandez-Gonzalez et al., 2004; Rinaudo and Schultz, 2004). In addition, the variability could be also acquired during the derivation of new lines, because different media are used in different settings until a stable line is established. However, in our setting all of these uncertainties were avoided, because all four lines were derived under identical conditions.
One other potential cause of variability could be caused by the timing of blastocyst processing. The human blastocyst normally arises around day 5 after fertilization, but the optimal timing for processing depends on the actual expansion status of the embryo. Thus, blastocysts are processed between days 5 and 8. During this critical time interval, the preimplantation blastocyst gradually shifts toward a postimplanting blastocyst, which changes the medium requirements as well as the expression profile of the embryo. In our hands, the best day for processing was day 6. Three of the cell lines (CHES-2, CHES-3, and CHES6) were processed at that time and only CHES-5 at day 7 (Table 1). An influence of the timing of blastocyst processing is not sufficiently plausible to explain the variability among CHES-2, CHES-3, and CHES-6, which were derived from blastocysts achieving a similar developmental stage after an identical culture period. We could not exclude such an influence in the differences between CHES5 versus the three other lines. However, authors of a recent study used embryos at different stages of development to derive ESC lines without causing a higher level of variability in gene expression profile among the lines (Giritharan et al., 2011). Thus, even in the case of CHES-5, it may still be possible that a different timing in blastocyst processing did not play a major role in the differences observed.
Our results confirm another recent study dedicated to compare cell lines generated under standardized conditions in which authors similarly observed a significant range of variability between cell lines (Lappalainen et al., 2010). One possible explanation could be that different cell lines are genetically determined to have different requirements or sensitivities to external factors used for maintenance of stemness in vitro. In accordance, we observed that medium composition greatly influenced the propensity of cells of a given line to remain pluripotent or to predifferentiate, but also changed transiently the extent of variation among different cell lines (A.C. Feutz, unpublished results). This suggests that slightly different compositions of the culture medium may be required to maintain stemness in each individual cell line.
Our hESC lines express differentiation markers at low levels characteristic of the three embryonic lineages, indicating spontaneous commitment during stem cells maintenance. Although the expression profiles of those differentiation markers are unique for each cell line, lines with the higher level of spontaneous commitment (CHES-2 and CHES-5) show striking similarities in expression profiles. That suggests that a common weakness may underlie their impaired ability to stay undifferentiated. The differences in gene expression profiles among different hESC lines were particularly pronounced in genes determining neuro-ectodermal differentiation, such as NeuroD1 and Pax6. This bias likely reflects the default status of neuro-ectodermal differentiation, which, in contrast to differentiation toward other lineages, does not require the presence of any specific inducers or cell interactions. As previously described (Lappalainen et al., 2010; Tavakoli et al., 2009), we observed that an increased tendency to spontaneously commit during stem cell maintenance correlates with an apparent increased efficiency to generate neurons when differentiation is allowed. However, while previous data have crudely described the divergent capacity of hESC lines to differentiate to neural cells, we found that our four hESC lines differentiate at similar rates and that the generation of more, or more mature, neurons after an identical time period simply reflects their distinct status already present at the onset of the differentiation process.
Furthermore, while previous data suggested that cell lines able to generate more neurons faster would be more suitable for clinical applications, our analysis of the differentiation process suggests that it may not necessarily be the case. Indeed, as shown in the most permissive situation, different levels of initial neural commitment were associated with clear abnormalities in the neural differentiation process. Following the timely evolution of expression of various markers during differentiation, we observed discordant behavior resulting from heterogeneity in the starting cell populations. For cell lines with the greater tendency to commit spontaneously, the progressing waves of ongoing differentiation of the already committed cells overlapped with those of newly committed cells. Such discordance is responsible for the observed lower ability to form rosettes in these populations, because rosette formation implies simultaneous acquisition of neuro-epithelial properties in neighboring cells. It may also explain the previous description of the different response to neuronal patterning factors of two similarly derived hESC lines, interpreted at that time as difference in differentiation potential (Wu et al., 2007). Indeed, the selective generation of a neuronal subtype in vitro, a prerequisite for a number of expected uses of hESCs, including in regenerative medicine, requires addition of regionalizing factors during a precise and narrow window of neuronal development (Gaspard and Vanderhaeghen, 2010; Okada et al., 2008). Treatment of heterogeneous cultures, with an increased proportion of cells at different stages of their development, would thus necessarily lower the ratio of the desired neuronal types or even bias the differentiation toward the generation of unwanted ones. Additional experiments are clearly needed to confirm that, at least in some cases, a higher level of neuron production correlates with increased difficulties to produce homogeneously specific neuron types.
Conclusions
The use of hESCs for drug discovery and toxicology requires cell lines with different genotypes but standardized differentiation behavior. The best strategy to achieve that goal may imply either selection of cell lines with closely related properties or control of the cells' tendency to predifferentiate spontaneously. In any case, the gene expression profile and the tendency to predifferentiate for each newly created hESC line should be well known and tested in a variety of different culture systems. The selection of a panel of cell lines with similar properties is clearly possible and straightforward. However, this strategy finds its limitation when a given cell line is not exchangeable, i.e., when a precise or compatible genotype is required, as in transplantation of tissue-engineered cells. That problem is even more pronounced when induced pluripotent stem cells (iPSC) lines are concerned. This indicates that a variety of different culture conditions may be required to maintain the stemness of stem cells derived from various sources.
Footnotes
Acknowledgments
We are grateful to both O. Hovatta and A. Stromberg (Karolinska Hospital, Stockholm, Sweden), who familiarized us with the handling of hESCs, and to M. Jaconi and A. Feki (Geneva University Hospitals) for supporting our initial research on hESCs in Switzerland. We also thank Mss. Susanne Grieshaber and Rita Epper, Institute for Pathology, University of Basel, for skillfully performing immunohistochemistry on the tumor sections. This research was funded by the Swiss National Fund [Joint Embryonic Stem Cell Project (JESP), B-FP-GV-6-0001-0001] and by the Swiss Center for Applied Human Toxicology (SCAHT).
Author Disclosure Statement
All authors declare no conflict of interest.
Author contributions were as follows: Oliver Sterthaus—conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; Anne-Catherine Feutz—conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; Hong Zhang—conception and design, data analysis and interpretation; Flurina Pletscher—data analysis and interpretation; Elisabeth Bruder—Data analysis and interpretation; Peter Miny—data analysis and interpretation Giandomenica Lezzi—provision of study material, data analysis and interpretation; Maria De Geyter—conception and design, provision of study material; Christian De Geyter—conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.