
Abstract
Abstract
Trisomy 21 is the most common chromosomal abnormality and is associated primarily with cardiovascular, hematological, and neurological complications. A robust patient-derived cellular model is necessary to investigate the pathophysiology of the syndrome because current animal models are limited and access to tissues from affected individuals is ethically challenging. We aimed to derive induced pluripotent stem cells (iPSCs) from trisomy 21 human mid-trimester amniotic fluid stem cells (AFSCs) and describe their hematopoietic and neurological characteristics. Human AFSCs collected from women undergoing prenatal diagnosis were selected for c-KIT+ and transduced with a Cre-lox–inducible polycistronic lentiviral vector encoding SOX2, OCT4, KLF-4, and c-MYC (50,000 cells at a multiplicity of infection (MOI) 1–5 for 72 h). The embryonic stem cell (ESC)-like properties of the AFSC-derived iPSCs were established in vitro by embryoid body formation and in vivo by teratoma formation in RAG2−/−, γ-chain−/−, C2−/− immunodeficient mice. Reprogrammed cells retained their cytogenetic signatures and differentiated into specialized hematopoietic and neural precursors detected by morphological assessment, immunostaining, and RT-PCR. Additionally, the iPSCs expressed all pluripotency markers upon multiple rounds of freeze-thawing. These findings are important in establishing a patient-specific cellular platform of trisomy 21 to study the pathophysiology of the aneuploidy and for future drug discovery.
Introduction
G
iPSCs have been generated from several postnatal somatic tissues, such as fibroblasts, keratinocytes, peripheral blood cells, neural stem cells, and hepatocytes (Aasen et al., 2008; Jeong et al., 2009; Liu et al., 2010; Seki et al., 2012; Takahashi et al., 2007). However, their reprogramming efficiency is considerably low, varying between 0.001% and 0.01%, depending on the source tissue. In addition, mutations accumulated during postnatal life may compromise their usage, and the tissue of origin may have an epigenetic influence on the ability of iPSCs to undergo directed differentiation (Kim et al., 2011; Mukherjee and Thrasher, 2011). Fetal cells are younger than postnatal cells and consequently are expected to carry fewer mutations, making them genetically more stable. They are also more primitive and less epigenetically modified than their adult counterparts (Kim et al., 2011; Li et al., 2009; Shaw et al., 2011; Ye et al., 2009). Indeed, fetal cells may be obtained as a by-product of invasive testing in pregnancies identified as being at increased risk of fetal abnormality at no additional risk to the patient and without the donor site morbidity that can accompany adult tissue biopsy (Cananzi et al., 2009; Shaw et al., 2011). Importantly, fetal cells isolated prenatally also provide access to diseases that lead to late miscarriage and stillbirth or that are lethal at birth, thus representing a unique source to study unknown or rare diseases (Li et al., 2009).
Among fetal sources, euploid human amniotic fluid cells (AFCs) have been successfully reprogrammed (Anchan et al., 2011; Liu et al., 2012; Moschidou et al., 2012a, 2012b; Wolfrum et al., 2010). AF stem cells (AFSCs) can be isolated during pregnancy following routine amniocentesis, during amnioreduction, or at delivery when cesarean section is performed (Shaw et al., 2011). AFSCs express some pluripotency markers, including OCT4A, are broadly multipotent (De Coppi et al., 2007), have high kinetics, and can be differentiated into mesodermal lineages (Bollini et al., 2011; Piccoli et al., 2012; Sedrakyan et al., 2012); however, they are not pluripotent, as seen by their inability to form teratomata when injected into immunodeficient mice (De Coppi et al., 2007).
Here we report the generation of disease-specific iPSC lines from human second-trimester aneuploid (trisomy 21) AFSCs using a Cre-excisable polycistronic lentiviral vector expressing the four Oct4, Klf4, Sox2, and c-Myc (OKSM) transcription factors (Sommer et al., 2009). Very recently, another study has demonstrated the generation of iPSCs from trisomy 21 AFSCs with subsequent neuronal differentiation (Lu et al., 2013). Here we explore the hematopoietic potential of human trisomy 21 AF-iPSCs that could have relevant implication for disease modeling in trisomy 21, where hematological malignancies are common.
Materials and Methods
Ethics statement
Pregnant women attending the Fetal Medicine Unit at University College London Hospital (UCLH) gave written consent for the study, which had ethical approval (NRES Committee London–Bentham. Study title: Amniotic fluid and placental stem cells. REC reference: 08/H0714/87). The mice were kept according to Home Office regulations and were euthanized by exposing them to CO2 covered by the approved Project Licence, number 70/7024. The Home Office Project License was reviewed by the UCL Animal Scientific Procedures Ethics Committee and the Royal Veterinary College Animal Scientific Procedures Ethics Committee, and also by the UK Home Office under the Animals (Scientific Procedures) Act 1986.
Isolation, culture, and characterization of AFSCs
Samples of AF (3–5 mL) were collected during ultrasound-guided amniocentesis in pregnant women at increased risk for trisomy 21 between 16 and 18 weeks of gestation. Fresh AF was filtered and c-KIT+ cells were isolated using monoclonal antibodies conjugated to MicroBeads (Miltenyi Biotec). Selected cells were grown in defined medium as previously reported (De Coppi et al., 2007). Fluroescence-activated cell sorting (FACS) analysis was performed on euploid and trisomy 21 AFSCs as described in Supplementary Data (Supplementary Data are available at www.liebertpub.com/cell/).
Derivation, culture, and characterization of AF-iPSCs
A total of 50,000 AFSCs were seeded on 35-mm culture plates, precoated with Matrigel (BD Biosciences). The cells were infected using a concentrated viral vector (see Supplementary Data) over a range of multiplicities of infection (MOI) varying between 1 and 5. At 72 h postinfection, the medium was replaced with human embryonic stem cell (ESC) medium (mTESR1, Stem Cell Technologies) supplemented with 0.5 μM PD0325901 and 2 μM SB431542 (InvivoGen). Within 1–2 weeks, compact colonies with ESC morphology appeared. The colonies were live-stained with fluorochrome-conjugated anti-SSEA4 antibody, picked up, and expanded when they reached 70% confluence using Collagenase Type IV (Stem Cell Technologies) to avoid single-cell suspension or, when desired, using TrypLE Express (Life Technologies) to obtain single-cell suspension in the presence of rho-kinase (ROCK) inhibitor (Y-27632, BioVision). Aliquots of AF-iPSCs were cryopreserved in liquid nitrogen using Stem Cells Keep (BioVerde, Inc), a vitrification solution that does not contain proteins, and dimethylsulfoxide (DMSO). For this purpose, semiconfluent T25 flasks containing trisomy 21 AF-iPSCs were mechanically detached into clumps (approximately 100 cells per clump) using Collagenase IV and frozen immediately in liquid nitrogen using Stem Cells Keep. In addition Rock Inhibitor (Y-27632) was added in the medium 1 h before cell harvesting and for 24 h after thawing to increase survival of single cells and smaller cell clumps (Baharvand et al., 2010).
Alkaline phosphatase (AP) activity was assayed using the AP kit from Sigma, in accordance with the manufacturer's instruction. AF-iPSC colonies were fixed with 4% paraformaldehyde/phosphate-buffered saline (PFA/PBS) for 15–20 min, permeabilized with 0.1% Triton X-100/PBS for 10 min, and blocked with 4% normal goat serum/PBS for 30 min (see Supplementary Data). Finally, quantitative real-time RT-PCR was performed as previously described (see Supplementary Data). The primers used are listed in Table S2.
Embryoid body formation
For embryoid body (EB) generation, AF-iPSCs were incubated with 1 mg/mL Collagenase type IV at 37°C for 15 min, dissociated into small clumps using a scraper, and cultured in suspension in ultra-low-attachment six-well microplates in differentiating medium containing Dulbecco's Modified Eagle Medium (DMEM)/F12 (SAFC Biosciences) supplemented with 20% ESC-qualified fetal calf serum (FCS; Invitrogen), 1% penicillin/streptomycin (Sigma-Aldrich), 1% nonessential amino acid (Sigma-Aldrich), 0.1 M β-mercaptoethanol (Invitrogen), and 1% Glutamax (Gibco, Invitrogen), for 10–15 days.
Teratoma formation assay
For generating teratomas, 75 μL of AF-iPSCs (1×106) resuspended in RPMI medium were added to 75 μL of Matrigel on ice (1:1, total volume 150 μL) and then injected subcutaneously into triple-knockout (RAG2−/−, γ-chain−/−, C2−/−) immunodeficient mice. Mice were observed for 8–10 weeks for the growth of teratomas. Teratomas were isolated, fixed in formalin, and embedded in paraffin. Hematoxylin & Eosin (H&E) staining (neural tube, ectoderm; gut epithelium, endoderm; bone and cartilage, mesoderm; squamous epithelium, endoderm; muscle and cartilage and adipocytes, mesoderm) and immunostaining for the markers of the three germ layers (α-actin, mesoderm; α-fetoprotein, endoderm, and β-tubulin, ectoderm) were performed (see Supplementary Data).
Hematopoietic differentiation
Hematopoietic differentiation of AF-iPSCs was performed using a three-step co-culture–based protocol adapted from Niwa et al. (2011) with modifications. First, OP9 stromal cells (104 cells/cm2) were seeded 3 days before the start of co-culture in α-minimum essential medium (α-MEM) supplemented with 20% FCS, 1% Glutamax, and 1% penicillin-streptomycin (OCM). On day 0, AF-iPSC colonies were seeded onto OP9 cells at a density of 103 cells/cm2 and maintained for 4 days in OCM supplemented with 50 ng/mL recombinant human bone morphogenetic protein-4 (BMP-4; Peprotech). On day 4, culture medium was replaced with OCM supplemented with 40 ng/mL vascular endothelial growth factor-165 (VEGF-165) and 50 ng/mL stem cell factor (SCF) (both from Peprotech), and the culture was maintained for 6 days with a medium change every other day. On day 11, the mixed cell population was sorted by flow cytometry to obtain a pure population of Lin− [fluorescein isothiocyanate (FITC) anti-human Lineage cocktail; BioLegend], CD34+ (APC anti-human CD34; BD-Pharmingen), and CD45− (PE/Cy5 anti-human CD45, BioLegend) hematopoietic progenitors. In the last differentiation stage, the sorted cells were cultured for 8 days in semisolid medium containing a cocktail of myeloid differentiation–promoting cytokines and growth factors (Methocult H4434, Stem Cell Technologies). Colony-forming units (CFUs) were harvested on day 8, and analyzed for CD45 expression through flow cytometry. For this step analysis, a CD34-FITC antibody (BD Pharmingen) was used. Cytospin preparations of cells extracted from CFUs were subjected to Wright–Giemsa staining and analyzed under an inverted microscope (Olympus 1X70).
Neural differentiation
AF-iPSC colonies were either passaged en bloc using Collagenase IV or detached using TrypLE Express to obtain a single-cell suspension, and 1×105 cells were plated onto Matrigel-coated six-well plates in mTESR1 medium (Stem Cell Technologies). The day after seeding, medium was changed to neural medium containing 1:1 DMEM/F12 (SAFC Biosciences) and Neurobasal (Gibco) medium plus the supplements 0.5% N2 (Gibco), 1% B27 (Gibco). AF-iPSCs were grown in neural medium for 6 days. A concentration of 5×10−5 (M) all-trans-retinoic acid (RA; Sigma R2625) was added to the N2B27 medium for other 8 days. Morphological assessment was based on neurite outgrowths and on the recognition of neural rosette with characteristic morphology and radial arrangement. Immunostaining for Nestin, PAX-6, and tubulin III (Tuj1) and RT-PCR (see Table S1) were conducted to show the presence of neural precursor cells.
Results
AF-iPSCs can be derived from AFSCs of fetuses with trisomy 21
AFSCs from fetuses with trisomy 21 (Fig. 1Ae) displayed similar morphology to AFSCs from euploid fetuses (Fig. 1Aa), but with different cell division kinetics (Fig. S1A). Trisomy 21 AFSCs showed slower growth than euploid AFSCs as evident from the growth curve (Fig. S1A). Some euploid and trisomy 21 AFSCs expressed pluripotency markers, SSEA-4 (Fig. S1B b, f), TRA-1-60 (Fig. S1B c,g), and TRA-1-81 (Fig. S1B d, h). FACS analysis showed expression of CD117, CD166, and SSEA-4 in both euploid and trisomy 21 AFSCs. Both types of cells were negative for CD31 and for the hematopoietic markers CD45 and CD34 (Fig. S1C, D). AF-iPSCs were subsequently derived from both euploid and trisomy 21 AFSCs using a polycistronic lentiviral OKSM vector at two different MOI values, i.e., 2 and 5. Transduced cells were subsequently expanded on Matrigel in mTESR1 under feeder-free conditions. AF-iPSC colonies from both euploid and aneuploidy fetuses were visible as early as 5 days postinfection (Fig. 1A b, f); the colonies increased in size by day 7 (Fig. 1A c, g), showing a typical human ESC morphology. To compare reprogramming efficiency, day 8 and day 14 AF-iPSC colonies were live-stained with fluorochrome-conjugated anti-SSEA4 antibody (Chan et al., 2009) and fluorescent colonies were counted. We observed that the number of SSEA-4+ colonies at days 8 (MOI=5) and 14 (MOI=2) after transduction was similar in both euploid and trisomy 21 AF-iPSCs (Fig. 1B, C), confirming that a lower MOI of 2 is sufficient to drive reprogramming of AFSCs, and that the process of reprogramming is unaffected by the aneuploid variation. To evaluate the expression of reprogramming lentivector in euploid and trisomy 21 AF-iPSCs, AF-iPSC colonies were expanded for at least five passages before being transfected with an expression plasmid encoding Cre-recombinase, and RT-PCR for the expression of reprogramming the lenti-vector was performed. As expected, there was no detectable expression of polycistronic transcript in cells treated with the Cre plasmid (Fig. 1D). Additionally, to demonstrate maintenance of endogenous expression of the critical pluripotency marker Nanog in the reprogrammed cells after Cre-mediated excision of the reprogramming cassette, we infected AF-iPSCs with a lentiviral vector expressing green fluorescent protein (GFP) driven from a Nanog-dependent promoter. There was no GFP expression in AFSCs due to the lack of endogenous Nanog. Both euploid and trisomy 21 Cre-excised AF-iPSCs, however, showed strong and sustainable GFP expression, confirming the presence of high-levels of endogenous Nanog (Fig. 1E).
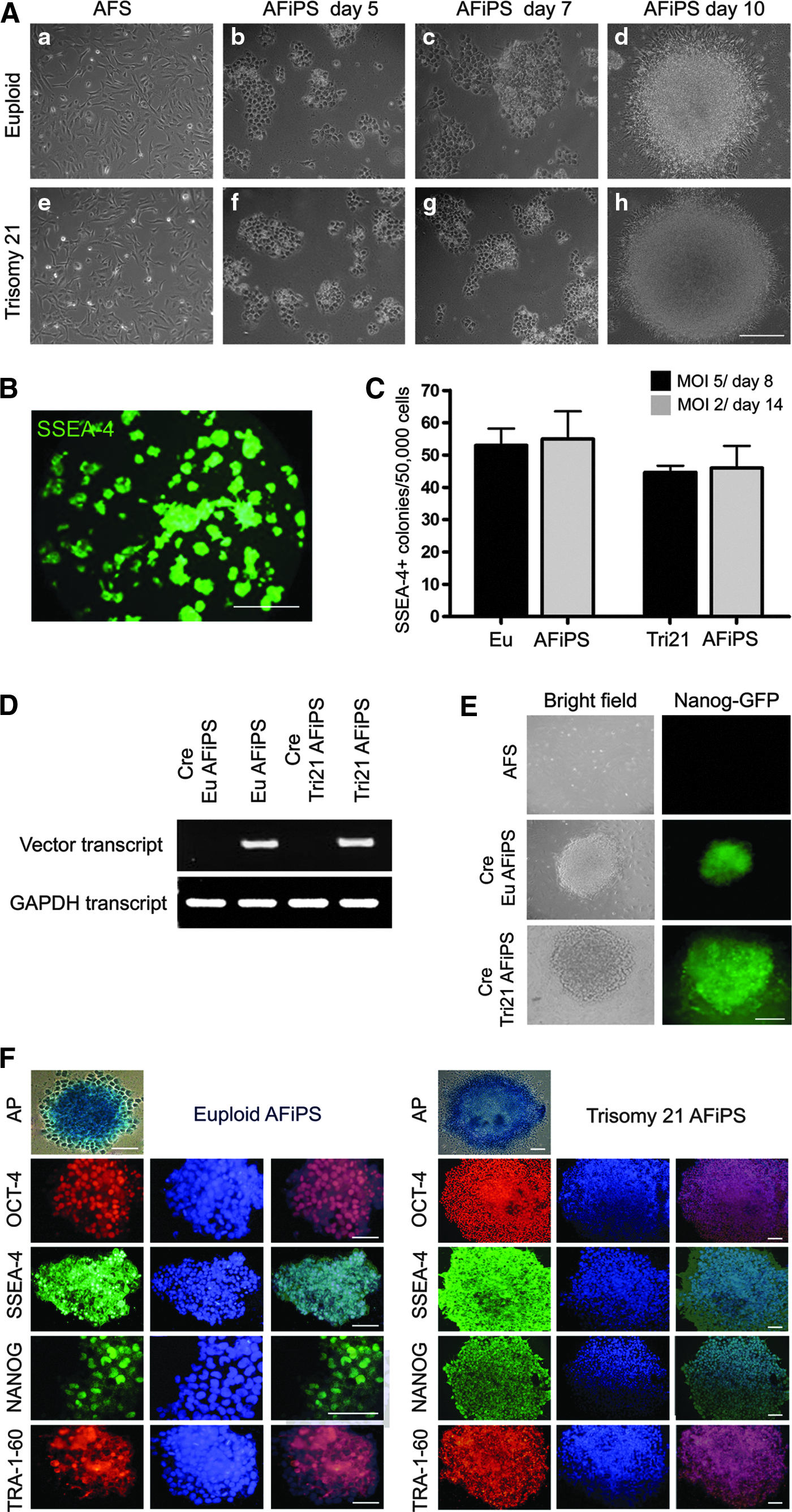
Characterization of euploid and trisomy 21 AF-iPSCs. (
After Cre-excision, normal and trisomy 21 AF-iPSCs were characterized for expression of pluripotency markers. Both populations stained positive for AP and were positive for OCT-3/4, SSEA-4, NANOG, and TRA-1-60 (Fig. 1F). The successful activation of endogenous pluripotency genes OCT-4, SOX-2, KLF-4, and c-MYC was verified by RT-PCR analysis (Table S1), which revealed that transgenic expression was absent in both euploid and trisomy 21 AF-iPSCs (Fig. 2A). Indeed endogenous expression of pluripotency markers was comparable to human ESCs, which were used as a positive control. When compared in a quantitative assay, both euploid and trisomy 21 AF-iPSCs expressed high levels of the pluripotency markers OCT-4 variant 1, SOX-2, KLF-4, c-MYC, and NANOG (Table S2). As expected, AFSCs, compared to their reprogrammed counterparts, showed moderate expression of OCT-4 variant 1, SOX-2, and NANOG and higher expression of KLF-4 and C-MYC (Fig. 2B). These results demonstrate that AFSCs from trisomy 21 fetuses can be efficiently reprogrammed.
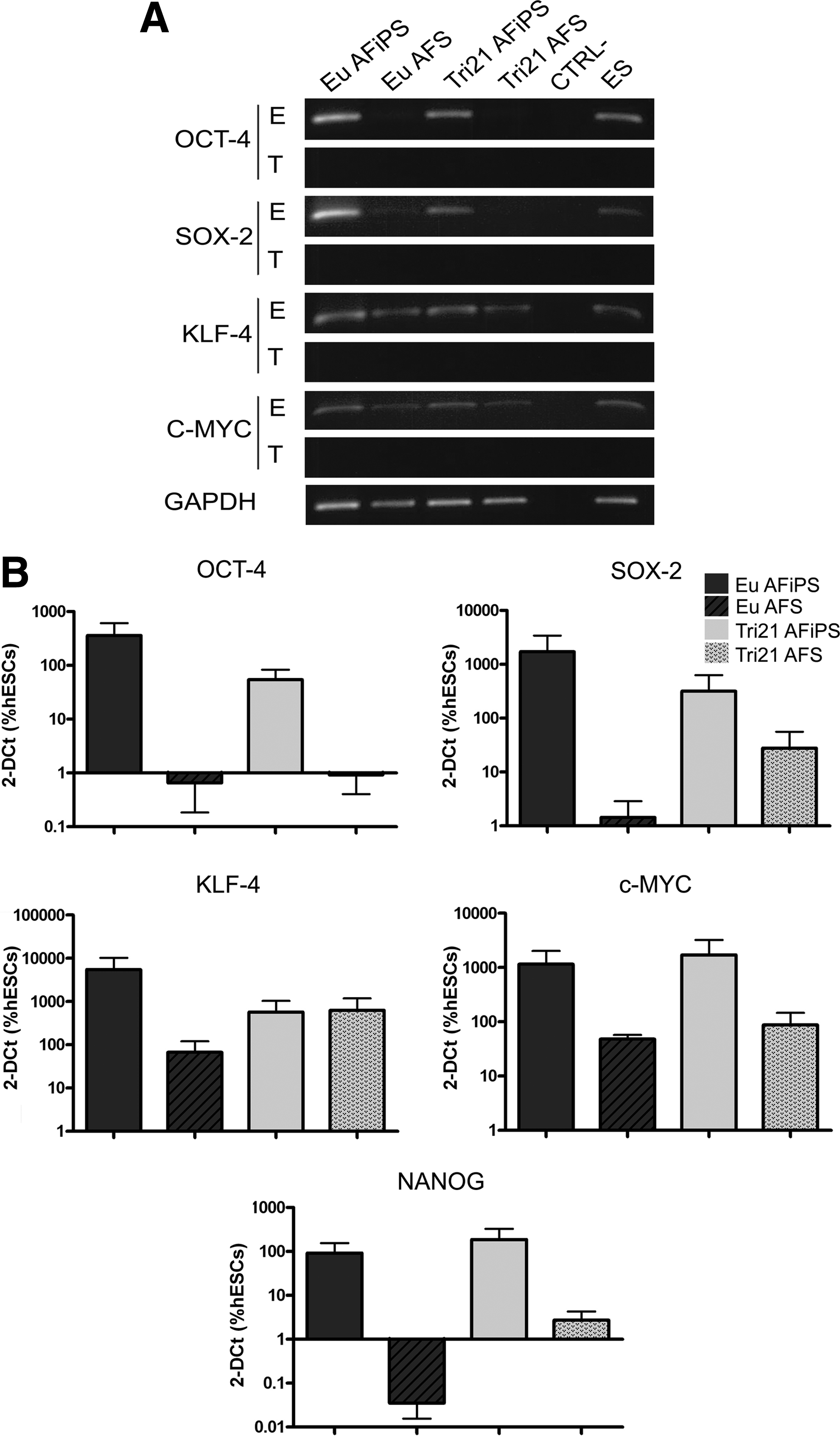
Molecular characterization of AF-iPSCs. (
Trisomy 21–derived AF-iPSCs maintain genetic signatures of the original source population
For the purposes of disease modeling and other preclinical investigations, it is essential that the cytogenetic features of source cells are maintained post-reprogramming (see karyotype and molecular cytogenetic analysis in Supplementary Data). As shown in Figure 3A, both euploid and trisomy 21 AF-iPSCs (propagated for at least 12 passages) showed the expected chromosomal complement and G-banding pattern. For trisomy 21 AF-iPSCs, the modal karyotype (a minimum of 19 metaphase spreads were evaluated) showed an abnormal female chromosomal constitution with the presence of an unbalanced 14;21 Robertsonian translocation (bottom image). The karyotype contains two normal chromosome 21s, one normal chromosome 14, and a derivative chromosome consisting of a fusion of the long arm of 14 and the long arm of 21 (indicated by arrowhead). All of these cells were therefore trisomic for the long arm of chromosome 21. The top image shows AF-iPSCs from normal AF cells that have maintained a normal female karyotype of 46,XX.

Euploid and trisomy 21 AF-iPSCs maintain genetic characteristics. (
For the Devyser Complete (Mix 1 and 2) and “in-house” QF-PCR mixes, all informative markers for chromosomes 13 and 18 (results not shown) and X and Y were consistent with a normal diploid complement (Fig. 3B). For chromosome 21, all informative markers [4/7 Devyser, 3/4 “in-house” (results not shown)] showed the presence of three alleles consistent with trisomy 21 [Fig. 3B and 3C (Devyser Complete Mix 1 only shown)]. Thus, the quantitative fluorescent polymerase chain reaction (QF-PCR) results were consistent with a female trisomy 21 patient for both the AFSC and AF-iPSC lines with no additional abnormalities detected in the transformed cell line. Additionally, both cell lines showed the same alleles present, which is consistent with the transformed cell line originating from the original cell line (Fig. 3B). The multiplex ligation-dependent probe amplification (MLPA) results showed increased peak ratios for both chromosome 21 probes in both telomeric probe mixes (P037 and P070) consistent with trisomy 21 for both the AFSC and AF-iPSC lines with no additional abnormalities detected in the transformed cell line (Fig. 3D).
Trisomy 21 AF-iPSCs form EBs
To assess in vitro pluripotency, both euploid and trisomy 21 AF-iPSCs were cultured in ultra-low-attachment plates for EB formation. When grown on nonadherent dishes in suspension in basic fibroblast growth factor (bFGF)-deficient culture medium, trisomy 21 AF-iPSCs assumed a compact spherical morphology by day 3, leading up to a more mature EB morphology by day 10 (Fig. 4A) with an efficiency of 80% compared to 100% for euploid AF-iPSCs (Fig. 4B). Confocal microscopy of trisomy 21 AF-iPSC EBs showed expression of markers from the three embryonic germ layers: α-actinin, β-tubulin (Tuj1), and α-fetoprotein (Fig. 4C). RT-PCR (Table S1) further confirmed expression of differentiation markers representative of all three germ layers. Euploid and trisomy 21 AF-iPSC EBs showed expression of α-fetoprotein (AFP) and GATA-4 (endoderm), RUNX-1 and Brachyury (mesoderm), and Nestin and PAX-6 (ectoderm); EBs from ESCs were used as positive controls (Fig. 4D). The absence of expression of these markers in AF-iPSCs further confirms the pluripotent state of these cells during their expansion in stemness conditions.
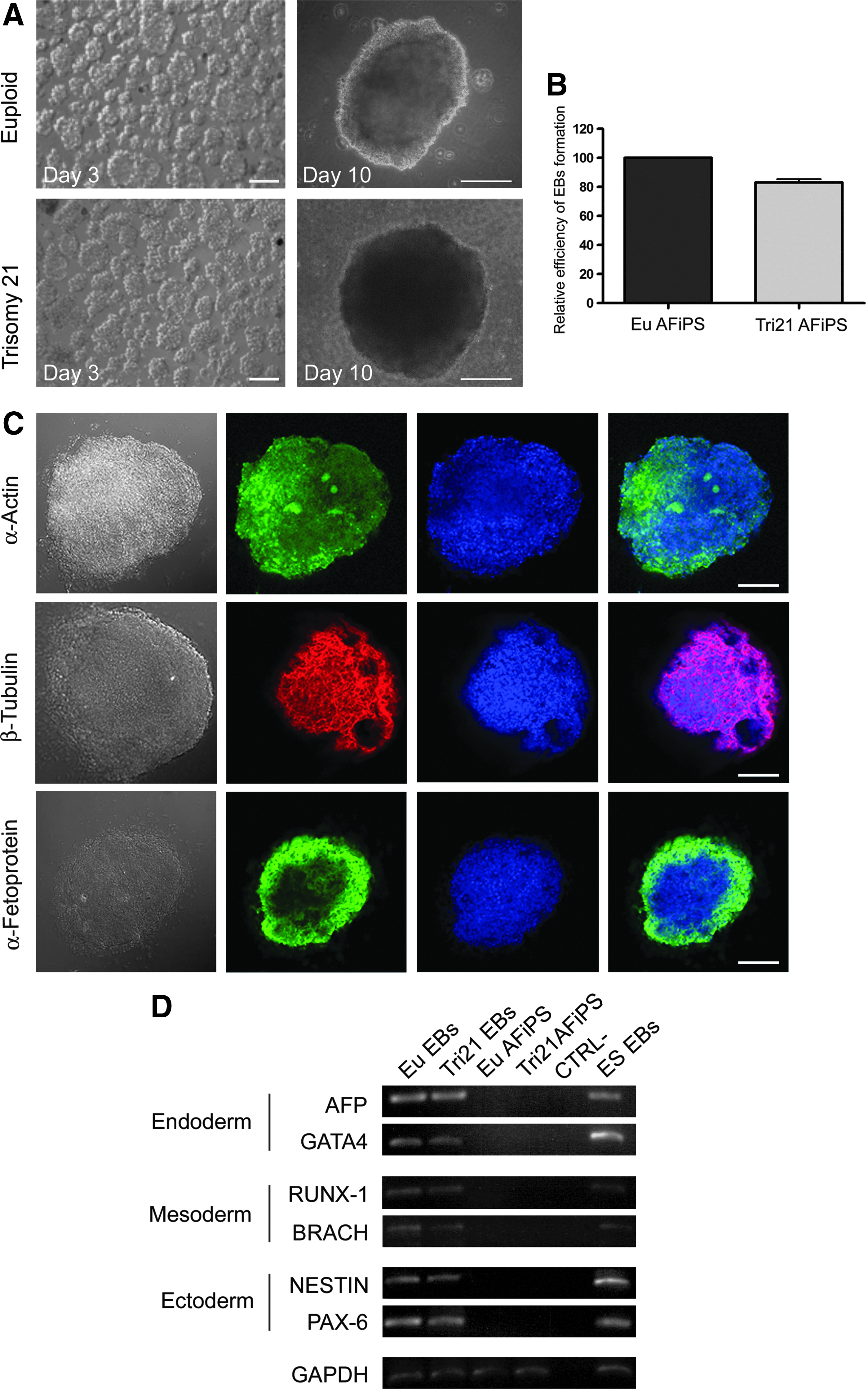
EBs generated from euploid and trisomy 21 AF-iPSCs. (
Trisomy 21–derived AF-iPSCs can generate teratomas in vivo
One of the stringent tests of pluripotency for human ESCs and iPSCs is their ability to generate teratomas in immunodeficient mice (Yamanaka, 2009). To further assess the in vivo capacity of AF-iPSCs to generate cell and tissue derivatives of the three germinal layers, normal and trisomy 21 AF-iPSCs were injected separately into triple-knockout (RAG2−/−, γ-chain−/−, C2−/−) immunodeficient mice. Approximately one million cells suspended in Matrigel were injected subcutaneously into the dorsal flank of immunodeficient mice, and tumors from the sites of injection were isolated between 8 and 10 weeks after injection (Fig. 5A). The AF-iPSC–derived tumors contained cells and tissue representatives of the three germ layers, as evident from the H&E staining of the paraffin-embedded sections showing the presence of neural tube (ectoderm), gut epithelium (endoderm), bone and cartilage (mesoderm), squamous epithelium (endoderm), muscle and cartilage, and adipocytes (mesoderm; Fig. 5B,C). Immunohistochemistry further confirmed the expression of the antigenic markers specific to each germinal layer: α-actin (mesoderm), AFP (endoderm), and β-tubulin (ectoderm) (Fig. 5B, C).
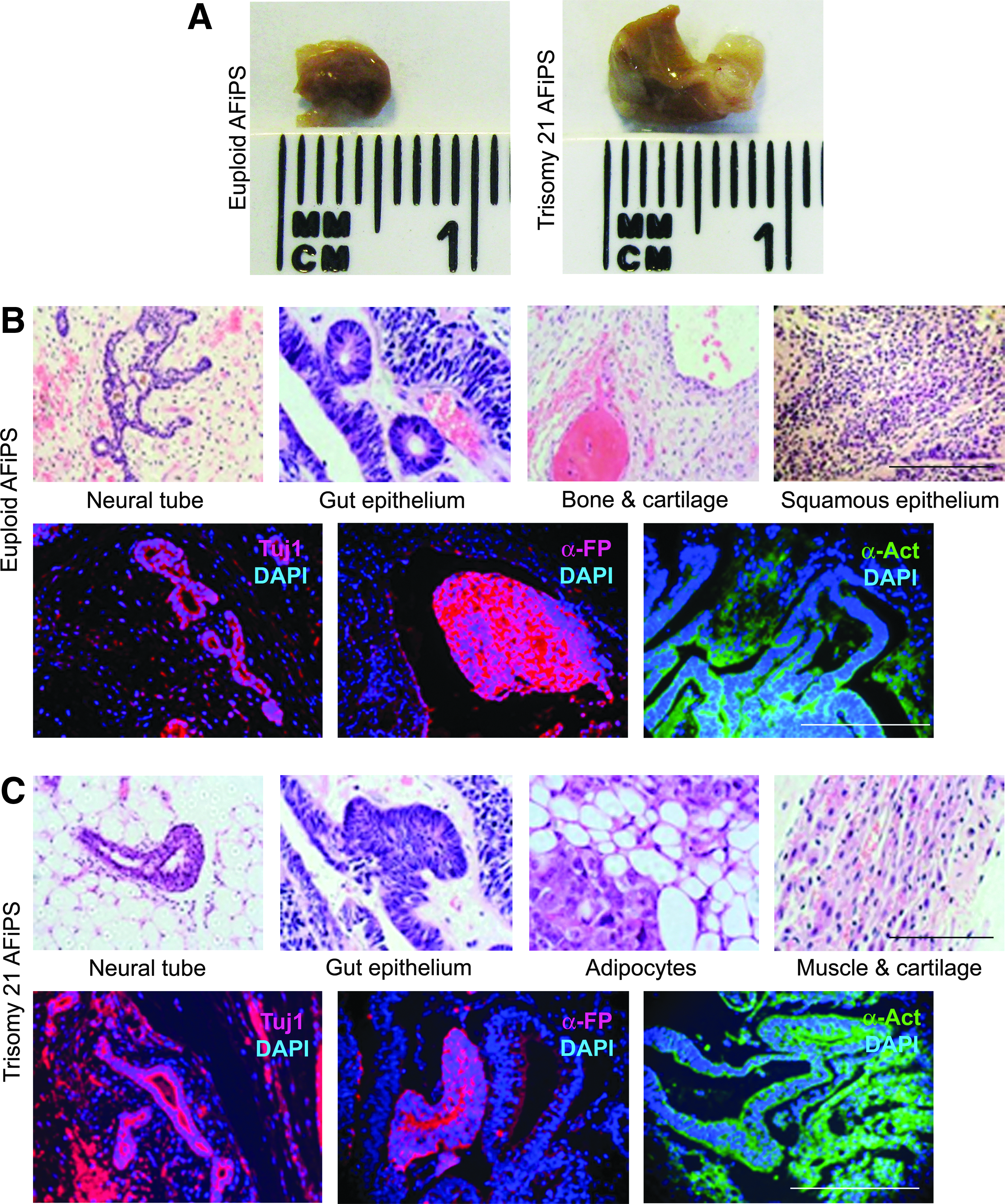
Teratoma generation by AF-iPSCs. (
AF-iPSCs can be frozen and banked
ESCs and iPSCs are very sensitive to cryopreservation, and the number of cells recovered after thawing is usually low (Baharvand et al., 2010). We observed that trisomy 21 AF-iPSCs maintained their morphology after thawing and grew in colonies similarly to fresh AF-iPSCs (Fig. S2A). Endogenous OCT-4, SOX-2, KLF-4, and C-MYC were demonstrated at RT-PCR (Fig. S2B, C). In addition, the pluripotency of cells was confirmed by immunofluorescence staining for the markers OCT-3/4, SSEA-4, TRA-1-81, and TRA-2-49. Finally, EBs were formed in selected conditions (Fig. S2D) and expressed markers of endodermal (AFP), mesodermal (Runx-1), and ectodermal (Pax-6) tissues (Fig. S2E). These results, which were obtained also with euploid AF-iPSCs (data not shown), suggest that cryobanking of trisomy 21 AF-iPSCs can be achieved.
AF-iPSCs can undergo directed differentiation in vitro into neural and hematopoietic lineages
AF-iPSCs were subjected to hematopoietic and neuronal differentiation protocols to estimate the feasibility of adopting them for future in-depth studies into the genetic disorders mentioned above.
Generation of hematopoietic progenitors and terminally differentiated myeloid cells from AF-iPSCs
Culturing AF-iPSCs on a confluent monolayer of OP9 cells in medium containing BMP4, and switching to VEGF-165- and SCF-containing medium resulted in the generation of hematopoietic progenitors. As shown in Figure 6B (top panel), after 10 days of selective differentiation, 96% of the euploid AF-iPSCs were lineage-negative (negative for CD3, CD14, CD19, CD20, CD56) and also negative for CD45 (pan-hematopoietic marker sans RBC). The same lineage negative population was found to contain around 4.3% CD34+ CD45− cells, which was sorted for enrichment and used in the terminal differentiation step (Fig. 6B, bottom panel). In this study, we employed a myeloid lineage–inducing cocktail for 8 days either in semisolid medium or liquid culture. At completion of the differentiation protocol (day 18), cells from the liquid culture were assayed by flow cytometry and were found to express CD45 uniformly with little or no expression of CD34 (Fig. 6C). At the same time, plates containing semisolid medium were assayed for colony formation [representative granulocyte-macrophage colony-forming unit (GM-CFU) colony shown in Fig. 6D], and CD45+CD34− cells isolated from the liquid culture as well as methylcellulose medium were used for cytospin preparations. Wright–Giemsa staining of the cells revealed myeloid cells, including granulocytes and monocytes (Fig. 6D), confirming terminal differentiation of the normal AF-iPSCs. We repeated the entire protocol employing two separately generated clones of trisomy 21 AF-iPSCs, and the gross outcome of hematopoietic induction is summarized in Figure 6E. On day 10, the percentage of CD34+ cells between the trisomy 21 AF-iPSC clones ranges between 7.8 and 8.3, comparable to what we observed for euploid AF-iPSCs (4.3%). At this stage, the percentage of cells expressing CD45 is also low (between 1.5% and 4%) and comparable to that of normal AF-iPSCs. However, upon completion of the differentiation protocol (day 18), we observed a low rate of conversion of CD34+CD45− cells to CD34−CD45+ immunophenotype in trisomy 21 AF-iPSCs (5–6%) compared with a rate of more than 90% when normal AF-iPSCs cells were used. Colony-forming cell (CFC) assays in semisolid culture of CD34+ cells from trisomy 21 AF-iPSC clones also failed to generate significant number of colonies (data not shown).
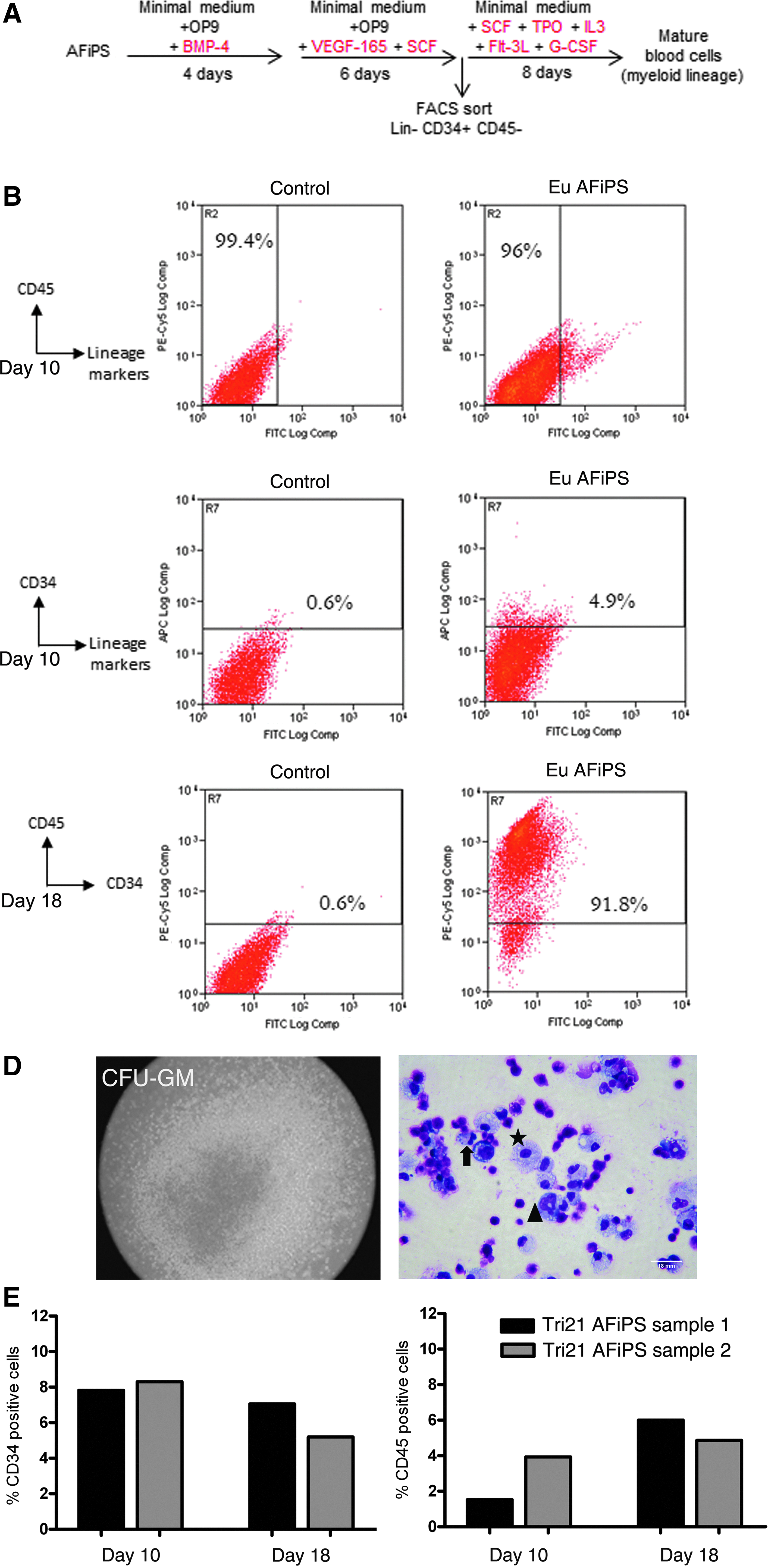
Hematopoietic differentiation of AF-iPSCs. (
Differentiation of AF-iPSCs into neural progenitors
Mature AF-iPSC clusters were passaged en bloc on Matrigel and switched from human (h) ESC medium to neural differentiation medium 24 h after seeding. After 6 days in differentiation medium, immunostaining revealed that both euploid and trisomy 21 AF-iPSCs are positive for the expression of Nestin and Pax-6 (Fig. 7A). Further treatment in retinoic acid–supplemented medium for 8 days gave rise to neural stem and progenitor cells that stained positive for neuronal differentiation marker β-tubulin III (Tuj1) in addition to Nestin (Fig. 7A). Control staining with anti-Oct4 and anti-Nanog antibodies failed to detect any expression of the pluripotency markers (data not shown). Neuronal stem cell differentiation was also inducible when trisomy 21 AF-iPSCs were dissociated into single-cell suspension and seeded onto Matrigel and induced using neural differentiation medium. At day 6, trisomy 21 AF-iPSC-neural induced cells were positive for Pax-6 (Fig. 7B) and showed the typical morphology of neural cells and rosette (Fig. 7C, D). RT-PCR further confirmed Nestin and Pax6 gene expression in differentiated trisomy 21 AF-iPSCs, whereas the undifferentiated cells were negative (Fig. 7E). Although we did not show the derivation of functional cortical neurons from these cells, we believe that the expression of early neuronal stem/progenitor markers like Pax6, Nestin, and Tuj1 combined with the formation of the neural rosettes and neuronal processes strongly indicate the ability of these cells to undergo terminal differentiation into cortical neurons, and therefore serve as a platform for anti-amyloid beta (Aβ) drug screening for the treatment of Alzheimer's disease.

Neural differentiation of AF-iPSCs (AFiPS). (
Discussion
In this study, we have demonstrated the hematopoietic and neuronal potential of iPSCs generated from trisomy 21 second-trimester human AFSCs. AF-iPSCs were generated using a polycistronic lentiviral vector encoding SOX2, OCT4, KLF4, and c-MYC with very good efficiency (≥0.1%). This efficiency should be compared to levels of iPSC generation of ∼0.002% from adult human fibroblasts (Sommer et al., 2009). The higher efficiency could be due to the fact that AFSCs, when compared to adult cells, may have advantages related to their epigenetic state that facilitate the acquisition of full pluripotency (Cananzi et al., 2009; Wolfrum et al., 2010). One study suggested that there were no apparent advantages for iPSC generation in selecting the cells for c-kit expression, although this was only investigated in mice (Anchan et al., 2011). Fully reprogrammed iPSCs could be generated from AFSCs within 10 days of transfection. Moreover, two different MOI values showed an equal efficiency of AF-iPSC generation, possibly due to the use of the chemical factors PD0325901 and SB431542 (Lin et al., 2009).
We particularly concentrated on the hematopoietic potential of AF-iPSCs. Up to 10% of neonates and adults with trisomy 21 suffer from a self-resolving transient myeloid disorder (TMD) within weeks after birth, and 20–30% of TMD patients progress to develop an acute megakaryocytic leukemia (AMKL) by age 1–4 years, which requires chemotherapeutic intervention. Although mutations in GATA1 have been implicated for this perturbed hematopoiesis, the exact mechanism of genetic predisposition to TMD and AMKL among trisomy 21 neonates is complex and poorly understood (Alford et al., 2011). Interestingly, AF-iPSCs may have an advantage to differentiate toward hematopoietic lineages over somatic cells because their freshly isolated AFSCs display a multilineage hematopoietic potential (Ditadi et al., 2009).
Previously, it has been shown by Ji and colleagues that OP9 stromal cells augment survival of the hematopoietic precursors and progenitors during hematopoietic differentiation from human ESCs (Ji et al., 2008). In the first step of induction of mesodermal progenitors, AF-iPSCs were cultured on a confluent monolayer of OP9 cells in medium containing BMP4, which has been previously shown to induce endo-mesodermal differentiation (Marshall et al., 2007; Nostro et al., 2008). Thereafter, the BMP4 medium was switched to VEGF-165- (ligand for KDR) and SCF- (ligand for CD117) containing medium to accelerate selective differentiation to hemangioblasts, as established in previous studies (Choi et al., 2009; Umeda et al., 2004). We showed a high level of terminal differentiation of the normal AF-iPSCs, but in only 5–6% of trisomy 21 AF-iPSCs were we able to demonstrate this. It is possible that downstream factors, such as mutations in the GATA1 gene of the trisomy 21 samples significantly affected lineage conversion. The difficulty of trisomy 21 iPSCs in vitro terminal hematopoietic differentiation may reflect a possible hematopoietic problem of trisomy 21 patients.
Previous studies have shown the generation of iPSCs from trisomy 21 fibroblasts (Mou et al., 2012; Park et al., 2008) and from AFSCs, with evidence of neuronal differentiation (Lu et al., 2013). Adults with trisomy 21 are at an increased risk of early-onset of dementia akin to patients with Alzheimer's disease. Trisomy 21 iPSCs from AFSCs would enhance our understanding of neuropathological consequences of trisomy 21, as it has been recently demonstrated that analysis of secreted Aβ42 and tau peptides (pathogenic peptides implicated in Alzheimer's disease) from short-term in vitro cultures of cortical neurons derived from trisomy 21 iPSCs can potentially provide a translational platform for drug targeting and toxicity screening (Shi et al., 2012; Yahata et al., 2011). Therefore, samples from individuals with trisomy 21 have the potential to provide a human cellular model for the in-depth study of these disease mechanisms to complement the existing animal models (Roubertoux and Carlier, 2010). Although human ESCs have been used to study aneuploid disorders (Biancotti et al., 2010), variability among individuals with trisomy 21 and penetrance can only be investigated from tissue biopsies that are difficult to access.
To be useful for studying mechanisms of disease, beside differentiation potential, it was important to demonstrate that AF-iPSCs generated from normal and trisomy 21 samples maintained the genetic characteristics of the respective parental cells. The pluripotency of AF-iPSCs were maintained upon freeze–thaw, providing support to the possibility of cryobanking of these cells to produce a tissue bank for researchers to access. Besides showing spontaneous differentiation through mesodermal, endodermal, and ectodermal lineages both in vitro (EBs) and in vivo (teratomas), we were successful in obtaining mature myeloid cells and neural precursors in vitro, confirming their usefulness for studying disease aetiology and for establishing drug screens.
Recently, we have demonstrated that reprogramming of first- (Moschidou et al., 2012a) and second- (Moschidou et al., 2012b) trimester c-KIT+ AF cells could be achieved without any genetic manipulation using only specific medium and valproic acid. We are currently studying the use of this technology to reprogram AFSCs from disease fetuses. Having established hematopoietic differentiation from AF-iPSCs, it could relevant to establish whether the role of trisomy 21 is similar to these cells, as very recently demonstrated by two elegant studies recently published on trisomy 21 iPSCs derived from different sources (Chou, et al., 2012; Maclean et al., 2012).
In conclusion, we have shown here that AF-iPSCs with hematopoietic and neuronal differentiation potential may be generated from human trisomy 21 AFSCs. Furthermore these AF-iPSCs can be cryopreserved and maintain their pluripotential. iPSCs derived from AF represent an ideal and renewable source of cells for in vitro modeling of congenital diseases. In addition to implications for cell banking, they may be useful to develop new treatments and for drug screening.
Footnotes
Acknowledgments
We would like to thank Dr. Terry Ballard of TDL Genetics for karyotype analysis; Dr. Ayad Eddaoudi of ICH Flow Cytometry Core Facility for help with FACS; and Rebecca West of ICH Pathology for preparing histology slides. P.D.C. is supported by the Great Ormond Street Hospital Children's Charity. M.C. thanks E.R.C. (Regenerative Therapy 269037). A.L.D. receives funding at UCLH/UCL via the Department of Health's NIHR Biomedical Research Centres funding scheme and an NIHR Clinical Senior Lectureship.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.