
Abstract
Abstract
DNA modifications, such as methylation and hydroxymethylation, are pivotal players in modulating gene expression, genomic imprinting, X-chromosome inactivation, and silencing repetitive sequences during embryonic development. Aberrant DNA modifications lead to embryonic and postnatal abnormalities and serious human diseases, such as cancer. Comprehensive genome-wide DNA methylation and hydroxymethylation studies provide a way to thoroughly understand normal development and to identify potential epigenetic mutations in human diseases. Here we established a working protocol for methylated DNA immunoprecipitation combined with next-generation sequencing [methylated DNA immunoprecipitation (MeDIP)-seq] for low starting amounts of genomic DNA. By using spike-in control DNA sets with standard cytosine, 5-methylcytosine (5mC), and 5-hydroxymethylcytosine (5hmC), we demonstrate the preferential binding of antibodies to 5mC and 5hmC, respectively. MeDIP-PCRs successfully targeted highly methylated genomic loci with starting genomic DNA as low as 1 ng. The enrichment efficiency declined for constant spiked-in controls but increased for endogenous methylated regions. A MeDIP-seq library was constructed starting with 1 ng of DNA, with the majority of fragments between 250 bp and 600 bp. The MeDIP-seq reads showed higher quality than the Input control. However, after being preprocessed by Cutadapt, MeDIP (97.53%) and Input (94.98%) reads showed comparable alignment rates. SeqMonk visualization tools indicated MeDIP-seq reads were less uniformly distributed across the genome than Input reads. Several commonly known unmethylated and methylated genomic loci showed consistent methylation patterns in the MeDIP-seq data. Thus, we provide proof-of-principle that MeDIP-seq technology is feasible to profile genome-wide DNA methylation in minute DNA samples, such as oocytes, early embryos, and human biopsies.
Introduction
DNA
DNA methylation is established and maintained by DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) that are essential for embryonic and postnatal development in mammals (Li et al., 1992; Okano et al., 1999). However, DNA methylation information is lost during standard molecular biology manipulations, such as molecular cloning in bacteria and PCRs, due to lack of maintenance of DNA methyltransferases. Therefore, several approaches have been proposed to preserve DNA methylation information and simultaneously transform it into quantitative and measurable signals (Laird, 2010). Of them, the most prevailing techniques include methylated DNA immunoprecipitation (MeDIP), bisulfite sequencing (BS-seq), and reduced representation bisulfite sequencing (RRBS). Combined with high-throughput, next-generation sequencing, these techniques have provided comprehensive genome-wide information regarding DNA methylation. In terms of the merit and bias of these methods, BS-seq and RRBS can generate a base-resolution DNA methylome, whereas MeDIP-seq can only generate relative enrichment of specific regions across the genome. Nevertheless, bisulfite treatment inevitably causes substantial DNA degradation and thus requires microgram levels of DNA Input. In addition, the bisulfite conversion efficiency is also a concern, even though many commercial kits claim more than 99% conversion. Special enzymes, such as uracil-tolerant DNA polymerase, are usually employed for high-throughput library preparation in BS-seq.
Affinity-enrichment methods such as MeDIP were initially combined with microarray technology to compare the genome-wide DNA methylation among samples (Weber et al., 2005). Subsequently, MeDIP was combined with a next-generation sequencing platform to investigate the DNA methylome in precious samples, such as bone marrow cells, mammalian oocytes, primordial germ cells, and preimplantation embryos. The MeDIP-seq protocol has been optimized to reach as low as 50 ng of DNA for starting materials (Taiwo et al., 2012). Borgel et al. performed MeDIP followed by whole-genome amplification and microarray hybridization on early-stage mouse embryos starting with 150 ng of DNA (Borgel et al., 2010). The sixth DNA modification 5-hydroxymethylcytosine (5hmC) can also be detected genome-wide by using a hydroxymethylated DNA immunoprecipitation (hMeDIP) procedure with a 5hmC-specific antibody (Ficz et al., 2011). Comparison between MeDIP-seq and hMeDIP-seq data helps better understand the distribution of 5-methylcytosine (5mC) and 5hmC signals on the same genomic loci across the whole genome. Recently, the MeDIP-seq method has been applied to detect other DNA modifications, such as 5-formylcytosine and 5-carboxylcytosine (Shen et al., 2013), thus illustrating the powerful advantages of MeDIP-Seq technology on genome-wide DNA modification studies.
The traditional MeDIP-seq requires microgram levels of starting DNA that may not be applied to minute DNA samples, such as mammalian oocytes and preimplantation embryos. Here we took advantage of next-generation sequencing technology to profile genome-wide DNA methylation by using 1 ng of genomic DNA from mouse embryonic stem cells (mESCs). After index adapter ligation, Input (without immunoprecipitation) and MeDIP samples were amplified by PCR for an Illumina HiSeq library preparation. Millions of reads were generated and then aligned to the mouse reference genome. The MeDIP-seq reads showed high alignment rate (97.53%) and were more piled up (indicating regions of DNA methylation) than Input reads across the whole genome.
Materials and Methods
mESC culture, embryoid body production, and neural differentiation
mESCs with a transgene for enhanced green fluorescent protein (eGFP) (B5 cell line) were cultured as adherent colonies in the presence of leukemia inhibitory factor (LIF; Chemicon), as previously described (Meyer et al., 2004). All chemicals are from Sigma (St. Louis, MO, USA) unless indicated otherwise. Briefly, undifferentiated ESCs were thawed and grown in gelatin-treated T25 tissue culture flasks in ESC growth medium plus LIF (ESGM; LIF at 1000 U/mL), including Dulbecco's modified Eagle medium (DMEM; Invitrogen), 10% newborn calf serum, 10% fetal bovine serum (FBS), nucleosides, and β-mercaptoethanol (1 mM). Typically, to maintain the undifferentiated state during proliferation, ESC cultures were refreshed every other day with ESGM plus fresh LIF and passaged from one to two T25 flasks. For future use, adherent ESC cultures were dissociated with 0.25% trypsin; the trypsin was inactivated with ESGM followed by a slow freeze to −80°C and transferred to liquid nitrogen for long-term storage. Otherwise, neural differentiation was achieved by using the 4−/4+ protocol that includes exposure to retinoic acid for neural induction (Bain et al., 1995). Briefly, the ESCs were cultured in ESC induction medium (ESIM; ESGM minus LIF and β-mercaptoethanol) in uncoated Petri dishes for 4 days and grown as suspended embryoid bodies (EBs). After 4 days in ESIM, the majority of ESCs formed floating spheres called EBs that had started neural differentiation. On day 4 of neural induction, the EBs were transferred into ESIM plus 500 nM all-trans retinoic acid (Sigma) and cultured for an additional 4 days to complete the neural induction phase. Finally, the EBs were collected, snap frozen in liquid nitrogen, and subsequently stored at −80°C until genomic DNA extraction.
Methylated DNA immunoprecipitation
Genomic DNA was extracted by using an AllPrep DNA/RNA Mini Kit (Qiagen) following the manufacturer's instructions. The DNA concentration was measured with a NanoDrop spectrophotometer (Thermo Scientific). The DNA was then randomly sheared into fragments with an average size of 400 bp by an M220 focused-ultrasonicator (Covaris). The MeDIP procedure was modified according to a standard protocol with low starting amounts of DNA (Borgel et al., 2012). To prevent DNA loss during the MeDIP procedure, special low DNA LoBind tubes (Eppendorf ) were used. Initially sonicated DNA (1 μg, 100 ng, 10 ng, and 1 ng) was diluted in 450 μL of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and then denatured at 95°C for 10 min. A portion of sonicated DNA was reserved as Input that did not undergo immunoprecipitation. Denatured DNA was placed on ice for 10 min and subsequently incubated with 5mC monoclonal antibody (Eurogentec, catalog no. BI-MECY-0100) or 5hmC polyclonal antibody (Active Motif, catalog no. 39769) for 2 h at 4°C with overhead shaking.
After linker ligation, standard cytosine, 5mC, and 5hmC DNA sets (Zymo Research, catalog no. D5405) were spiked into genomic DNA as controls as per the manufacturer's protocol (Ito et al., 2010; Wu et al., 2011). According to the manufacturer, virtually 100% of the DNA controls are converted to 5mC or 5hmC. DNA controls were isolated with Dynabeads Protein G (Invitrogen, catalog no. 10003D) by prewashing with 800 μL of phosphate-buffered saline (PBS)/0.1% bovine serum albumin (BSA) three times by using a magnetic rack and added to the immunoprecipitation samples. The Dynabeads were incubated with immunoprecipitation samples for 2 h at 4°C with overhead shaking. The Dynabeads bound by 5mC or 5hmC antibodies were then trapped by a magnetic rack and washed three times by using 1× immunoprecipitation buffer [10 mM sodium phosphate (pH 7.0), 0.14 M NaCl, and 0.05% Triton X-100] at room temperature with overhead shaking. The protein–DNA complex [DNA/Dynabeads/immunoglobulin G (IgG)] was subsequently digested with proteinase K (New England Biolabs, catalog no. P8102S) for 2 h at 50°C with shaking. The immunoprecipitated DNA was extracted by phenol/chloroform and precipitated by ethanol. The DNA pellet was then resuspended in EB buffer [10 mM Tris-HCl (pH 8.5)].
Real-time quantitative PCR
End-point PCR and real-time quantitative (q) PCR using immunoprecipitation and Input DNA were performed by using primers previously published (Mohn et al., 2009). Intracisternal A particle (IAP) was selected as a positive control (hypermethylated) and the promoters of Gapdh and β-actin were used as negative controls (hypomethylated) for mESC DNA. Spike-in control primers are listed in Table 1. Real-time qPCR was performed by using iQ™ SYBR Green Supermix in an iCycler IQ Single-Color Real Time PCR Detection System (Bio-Rad). Melting curves were generated following real- time PCR to assess the specificity of the amplicons. The copy numbers of initial template were analyzed by a relative standard curve method. Gradient dilutions (1/10×) of Input DNA were used to create standard curves. The qPCR data were obtained from three independent biological and two technical replicates and analyzed statistically by one-way analysis of variance (ANOVA).
MeDIP, methylated DNA immunoprecipitation; qPCR, quantitative PCR.
Illumina library preparation and high-throughput sequencing
The MeDIP protocol involves denaturation and generates single-stranded DNA, thus Illumina (San Diego, CA) adaptors must be added before immunoprecipitation. Illumina library preparation was performed by using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, catalog no. E7370S). Briefly, sonicated DNA (1 ng) was end repaired into dA-tailed fragments. Then the NEBNext indexed adaptor was ligated into sonicated DNA fragments. Adaptor-ligated DNA was further cleaned up by AMPure XP beads (Beckman Coulter, Inc., catalog no. A63881) and then subjected to the MeDIP procedure as described above. The Input DNA was linked to different index primers. After immunoprecipitation, adaptor-ligated DNA was extracted by phenol/chloroform and precipitated by ethanol. The antibody-enriched DNA was subsequently amplified by using an index primer and universal PCR primer provided in NEB Multiplex Oligos for Illumina (NEB, catalog no. E7500). For Input sample, 18 cycles were run, whereas 20 cycles were performed for MeDIP samples. The PCR products were then purified by AMPure XP beads and eluted with TE buffer. The library concentration was measured by a Qubit Fluorometric Quantitation system (Life Technologies), and size distribution was analyzed by a Fragment Analyzer™ Automated CE System (Advanced Analytical Technologies). The qualified libraries were then uploaded onto an Illumina HiSeq 2000 platform at the DNA Core (University of Missouri-Columbia) for ultradeep sequencing with single reads of 100 bases. Input control and MeDIP-seq data were submitted to the NCBI Sequence Read Archive (SRA) and are available under accession number SAMN02687582.
Reads alignment and data analysis
Raw single-read FASTQ data were first analyzed with FastQC Version 0.10.1 (www.bioinformatics.babraham.ac.uk/projects/fastqc/) for general read quality assessment. Then the FASTQ reads were preprocessed with Cutadapt (http://code.google.com/p/cutadapt/) to remove any adaptor and primer contamination. The trimmed data were then aligned to the mouse GRCm38 genome assembly via Bowtie2 short-read aligner (Langmead and Salzberg, 2012). (http://bowtie-bio.sourceforge.net/bowtie2/faq.shtml). Peak detection, visualization, and analysis of the aligned data were performed using SeqMonk (www.bioinformatics.babraham.ac.uk/projects/seqmonk/). “Probes” were defined in SeqMonk as genomic loci where quantitative measurements of read counts could be evaluated. The promoter region of each annotated gene was defined as being located between −1,000 bp and +1,000 bp flanking the transcription start site (TSS).
Results
The specificity of 5mC and 5hmC antibodies
To test the specificity of 5mC antibody, we spiked standard DNA (25 pg) sets containing 100% unmethylated cytosine (5C), 5mC, or 5hmC into mESC genomic DNA before immunoprecipitation. The standard DNA has three sets of linear double-stranded (ds) DNA (897 bp) that have the same sequence. All the cytosines in standard DNA sets are either 100% unmodified (5C), methylated (5mC), or hydroxymethylated (5hmC). The antibodies against 5mC (Eurogentec) and 5hmC (Active Motif ) were selected based on their performance in other laboratories (Ficz et al., 2011; Weber et al., 2005; Wu et al., 2011). End-point PCR was performed after immunoprecipitation by using antibodies against 5mC and 5hmC, respectively.
As shown in Figure 1A, 5mC was relatively enriched in MeDIP by using 5mC antibody, whereas 5hmC was highly pulled down in hMeDIP by using 5hmC antibody compared to other DNA modifications. Two independent regions of control DNA sets were selected and consistent data were obtained. Subsequently, real-time qPCR was performed to quantitate the relative enrichment of 5C, 5mC, and 5hmC versus Input after the MeDIP and hMeDIP procedure, respectively. For MeDIP, the 5mC antibody showed preferential enrichment over 5C and 5hmC (Fig. 1B). Likewise, the 5hmC antibody exhibited more than 100 times enrichment versus 5C and 5mC (Fig. 1C). Collectively, these data indicate the high specificity of 5mC and 5hmC antibodies for immunoprecipitation.
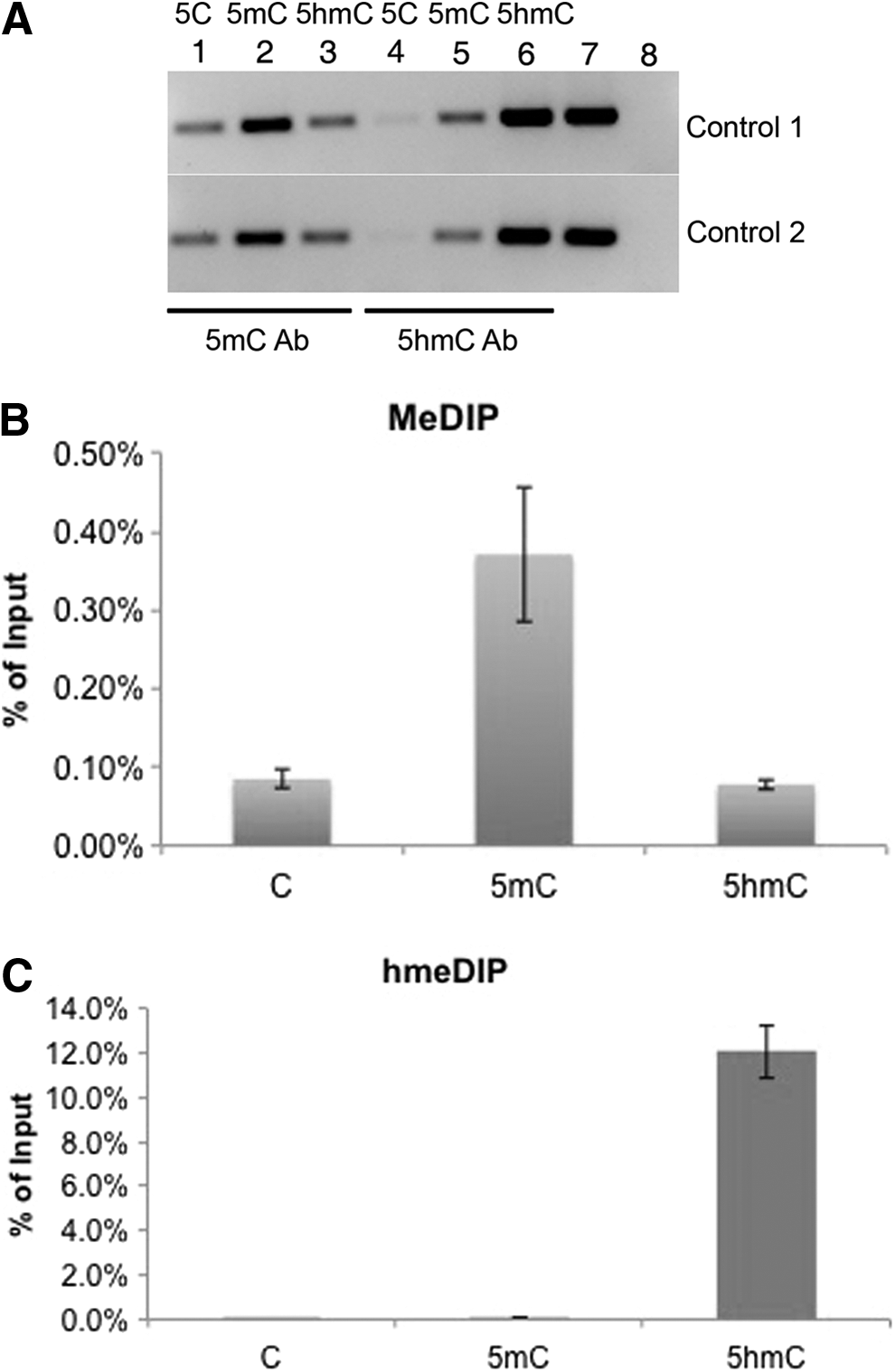
Specificity of 5mC and 5hmC antibodies. (
MeDIP-PCRs on representative genomic loci
Next we tested our MeDIP procedure on representative genomic loci. We used 100 ng, 10 ng, and 1 ng of mESCs or mEB DNA (data for mEB are not shown, but were similar to the mESCs) as starting material. For unmethylated controls, such as the promoters of Gapdh and β-actin, we did not observe bands after (h)MeDIP-PCR (Fig. 2A). Similarly, for intermediate methylated controls, such as differentially methylated region (DMR) in Igf2/H19, we did not see any band either, probably because of moderate methylation level and starting DNA (Fig. 2A). In fact, the DMR of Igf2/H19 has lower than 50% the methylation level in mESCs due to the global hypomethylation state of these genes (Hayashi et al., 2012; Leitch et al., 2013). In contrast, for a highly methylated region IAP, we observed abundant PCR product, even in 1 ng DNA samples for MeDIP, despite smaller amounts of PCR product in hMeDIP samples (Fig. 2A). That is reasonable because 5hmC only accounts for 5–10% of total 5mC in mESCs (Pastor et al., 2013). In addition, for constant amounts of spike-in controls, the enrichment efficiency was reduced with the decrease of starting DNA amounts (100 ng to 1 ng) for both MeDIP and hMeDIP (Fig. 2B). In contrast, for the endogenous highly methylated region (IAP), enrichment efficiency versus Input seemed to be higher in low DNA amount (1 ng; Fig. 2C). Therefore, the MeDIP procedure by using 1 ng of DNA is practical for discrete genomic regions and might also be applied to genome-wide DNA methylation studies.
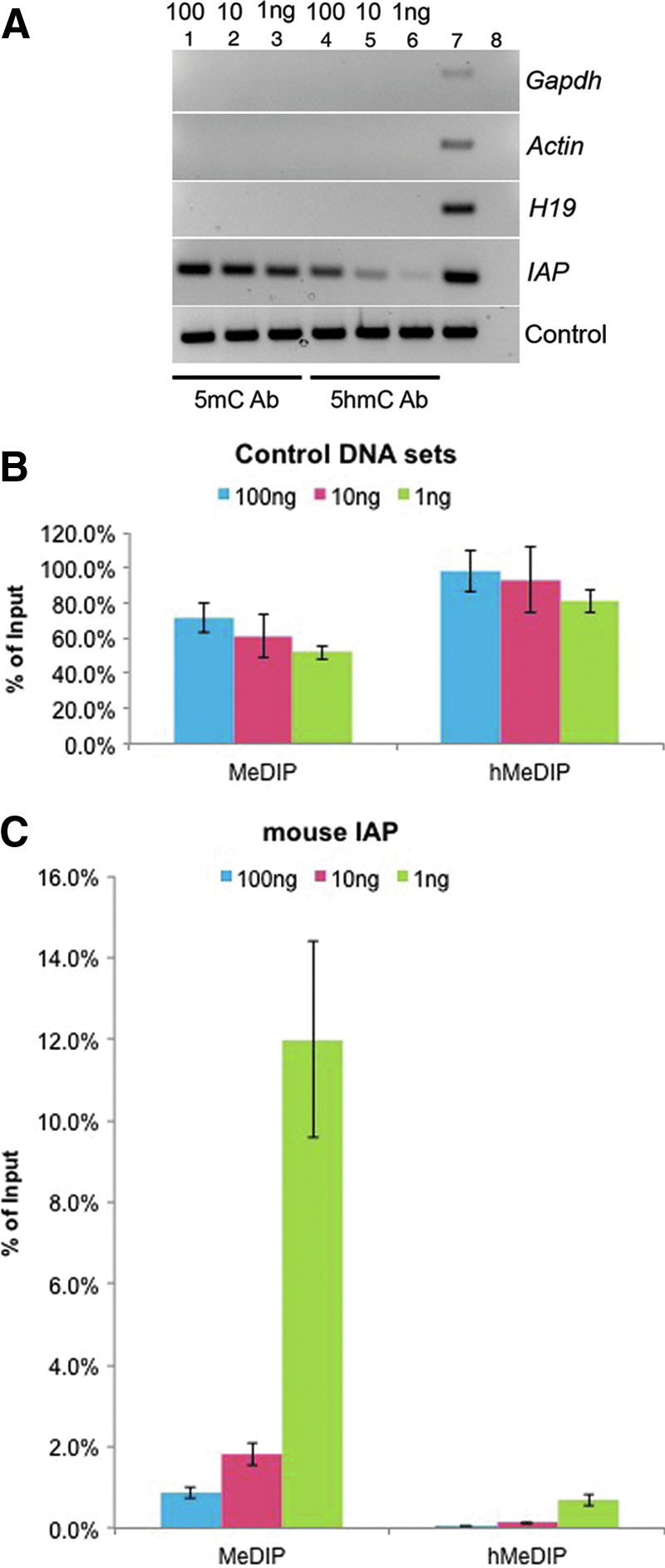
MeDIP/hmeDIP enrichment efficiency in exogenous and endogenous DNA by using a low amount of starting DNA. (
The quality of sequencing library by using 1 ng of DNA
To take the advantage of Illumina high-throughput sequencing HiSeq 2000 platform, index adaptors were added before immunoprecipitation of 1 ng of DNA for library preparation. After PCR amplification by using universal and index primers and clean up, the size distribution was analyzed. The majority of MeDIP-seq library fragments were between 250 bp and 600 bp, which was ideal for the downstream high-throughput sequencing (Fig, 3A). The MeDIP procedure removed adaptor contamination and shifted the average size of fragments when compared to the Input library without any enrichment. The Input fragments were distributed with a wider range of sizes, and primer/adapter sequences were obvious with peaks of 127 bp and 140 bp (Fig. 3B). Note that the MeDIP library required two more cycles of PCR than Input because the immunoprecipitation procedure caused about 90% loss of starting DNA.
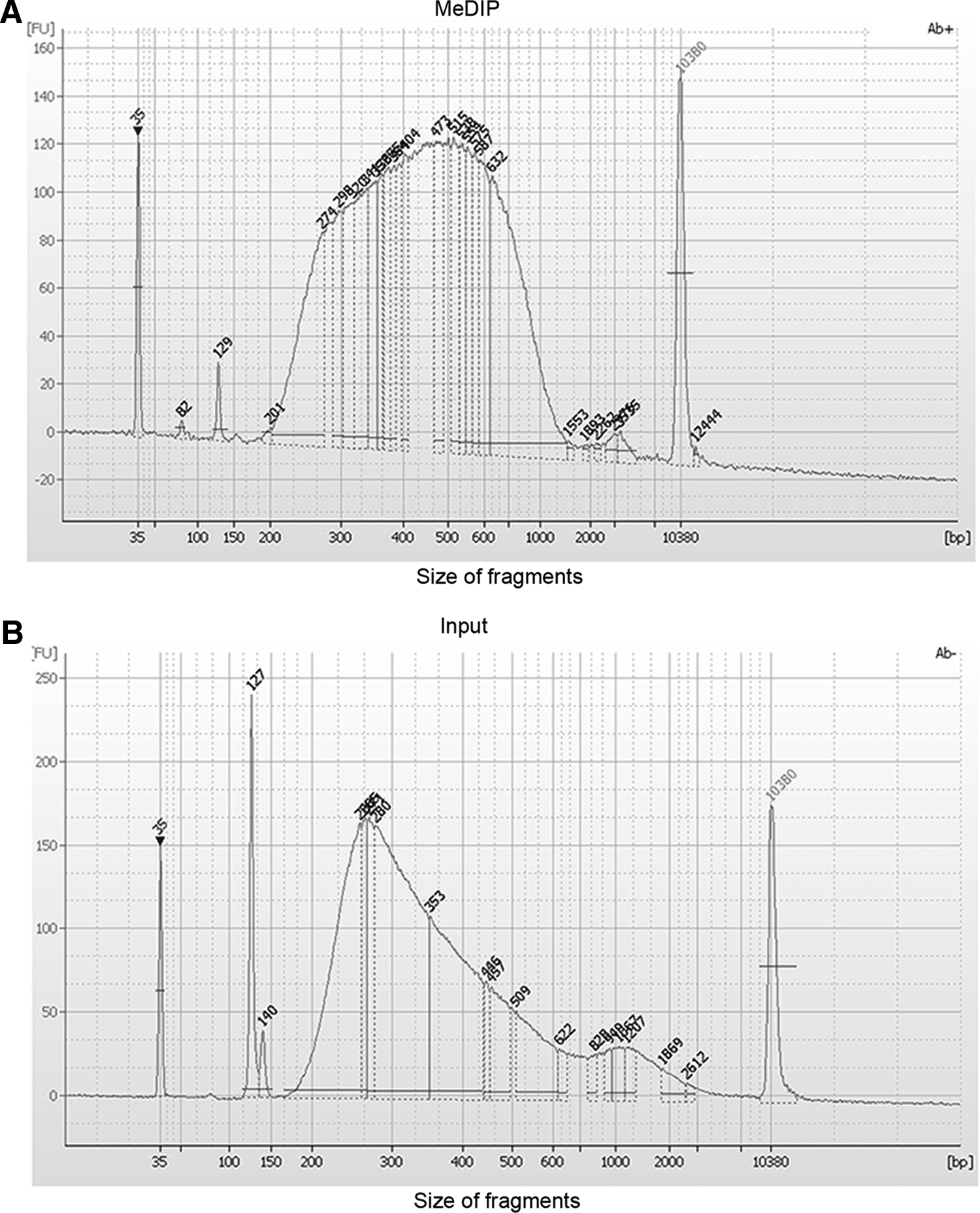
Fragmentation analysis of input and MeDIP-seq library by using 1 ng of DNA. The majority of MeDIP library fragments were between 250 and 600 bp after 5mC antibody enrichment (
Genome-wide DNA methylation profile by MeDIP-seq starting with 1 ng of DNA
The raw Illumina 100-bp single-read data (FASTQ) were analyzed with FastQC (v. 0.10.1) for quality assessment. Overall, MeDIP reads had higher-quality scores than Input reads. Before any processing, MeDIP read-quality scores declined at about 90 bases per read (Fig. 4C), whereas Input reads dramatically lost quality after 60 bases per read (Fig. 4A). The read-quality difference may be due to the disparate library quality between the Input and MeDIP samples, because the immunoprecipitation procedure could inevitably remove possible adapter and primer contamination and simultaneously enrich 200- to 500-bp fragments. After trimming by Cutadapt to remove any adapter and primers, the preprocessed MeDIP and Input reads showed comparable quality scores across all bases within 100 bp (Fig. 4B, D). As shown in Table 2, MeDIP reads had a 97.53% overall alignment rate, of which 64.66% reads were aligned exactly one time and 32.86% reads aligned more than one time. The mapped Input reads showed 94.98% alignment rate, of which 69.45% reads were aligned exactly one time and 25.53% were aligned more than one time. The comparable overall alignment rates (97.53% versus 94.98%) between Input and MeDIP indicate the high quality of these trimmed single reads.
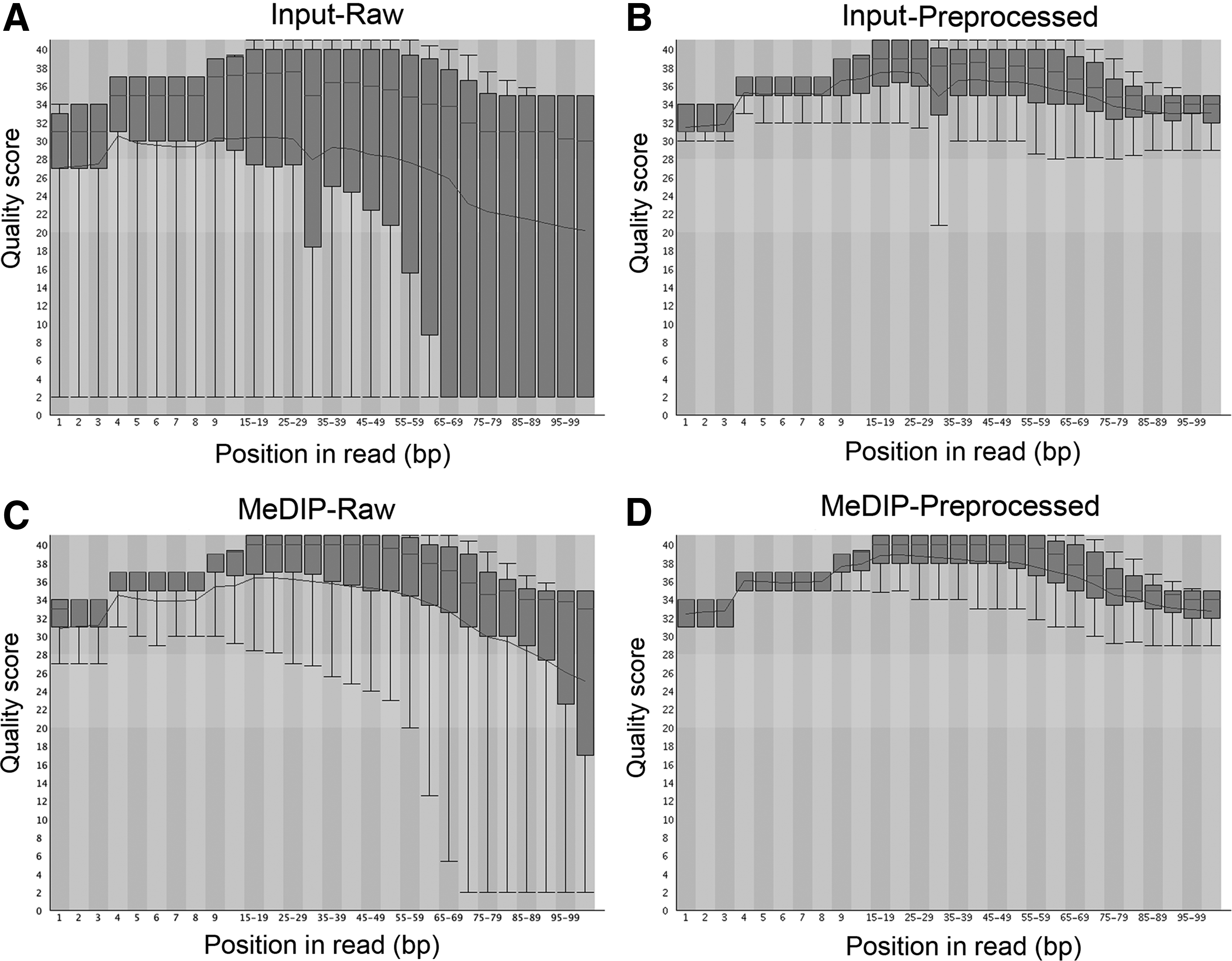
Quality assessment of MeDIP-seq and input reads by FastQC. The input read quality dropped dramatically after 60 bp per read (
MeDIP, methylated DNA immunoprecipitation.
By using SeqMonk built-in visualization tools, we observed that MeDIP-seq reads were discrete and piled up with gaps in between at selective genomic loci (Fig. 5A, bottom panel). In contrast, the Input reads were continuously and randomly distributed across the genome (Fig. 5A, upper panel), implying the successful enrichment of methylated DNA by the 5mC antibody. Occasionally, MeDIP reads were packed at selective locations, resulting in significant “probe” value bars. These probe value bars were usually located at highly methylated regions, such as gene bodies of Tet1 and Tet2 (Table 3). In contrast, probe values were seldom seen in hypomethylated regions, such as the promoter regions of housekeeping genes (Ywhag) and pluripotency genes (Nanog, Sox2, Pou5f1, and Stat3) that were highly expressed in mESCs, although there was substantial methylation within the gene bodies (Ywhag and Stat3) (Table 2). For example, there were no reads enriched in the promoter of Nanog in the MeDIP sample, but a substantial number of reads were located in the Input (Fig. 5C).
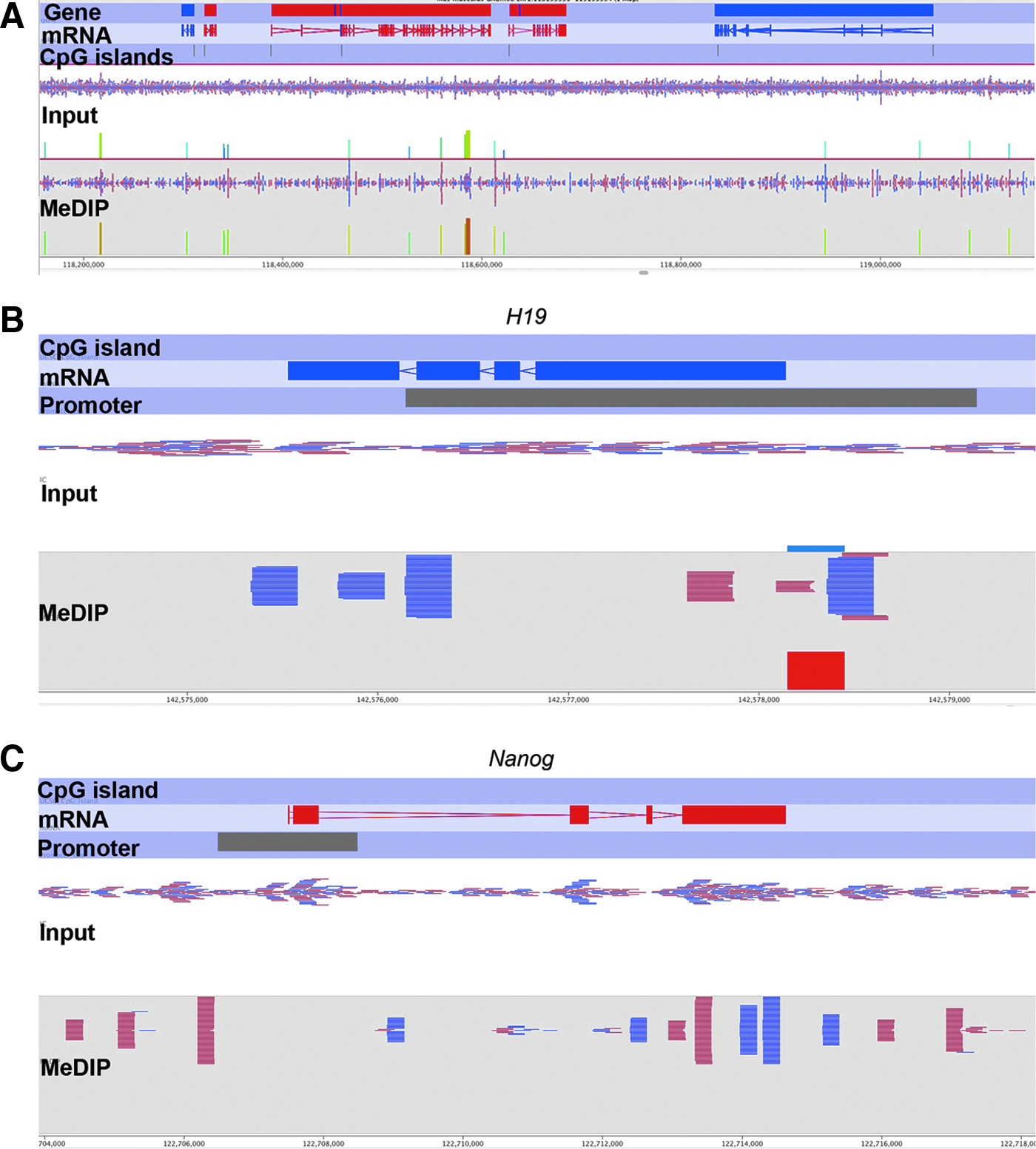
Visualization of MeDIP-seq data by SeqMonk in selective genomic loci. (
The higher enrichment fold, the more methylation in defined probes.
ESCs, embryonic stem cells; MeDIP, methylated DNA immunoprecipitation.
Lineage-specific genes were methylated either in the gene body (Meis1) or in the promoter (Hoxb1) to prevent their precocious expression in undifferentiated ESCs (Jaenisch and Young, 2008). For imprinted genes, the H19 promoter was highly methylated (Table 4), corresponding to the allele-specific methylation in the Igf2/H19 locus (Bartolomei and Ferguson-Smith, 2011). The enrichment was reflected by the probe value bars and piled reads seen in the promoter of H19 gene (Fig. 5B). Likewise, the Igf2r gene body and promoter region were both moderately methylated, which may contribute to its imprinted expression solely from maternal allele (Wutz et al., 1997). (Supplementary Data of the entire data set are available at http://animalsciences.missouri.edu/faculty/prather/ and www.liebertonline.com/cell/.) Together, these data support the idea that MeDIP-seq starting with 1 ng of DNA can generate a reliable genome-wide DNA methylation profile.
The higher enrichment fold, the more methylation in defined probes. Note that the following promoters (Nanog, Pou5f1, Ywhag, Sox2, and Stat3) were unmethylated and thus not listed in the table.
ESCs, embryonic stem cells; MeDIP, methylated DNA immunoprecipitation.
Discussion
In this study, we have established a working MeDIP protocol by using 1 ng of DNA as the starting material to profile genome-wide DNA methylation. End-point PCR products followed by MeDIP can be visualized at highly methylated endogenous regions (e.g., IAP). The enrichment efficiency was increased for endogenous loci but was reduced for constant spike-in controls with the decreasing amounts of starting DNA. Piled-up peaks were observed discretely across the genome in our MeDIP sample, whereas Input reads were continuous and randomly distributed. Known methylation patterns were also recapitulated in our MeDIP-seq data. The ability to accurately detect global DNA methylation patterns in minute samples will be of tremendous value when dealing with samples that contain very small amounts of DNA, such as oocytes, early embryos, and human biopsies.
Affinity-based methods (MeDIP/hMeDIP) coupled with next-generation sequencing can be used to profile genome-wide DNA methylation patterns with the resolution of several hundred base pairs and at a competitive cost. To achieve comprehensive genomic coverage and a viable compromise between sequencing breadth and depth, MeDIP-seq requires a range of 30–60 million reads per sample, whereas whole-genome BS-seq should sequence over a billion reads per sample (Bock et al., 2010). Although BS-seq and RRBS can generate single-nucleotide resolution, sodium bisulfite may also destroy the integrity of DNA and introduce possible mutations. In addition, bisulfite conversion is usually less efficient or variable for cytosines at non-CpG positions (Warnecke et al., 2002), which may cause a biased methylation profile at non-CpG sites. In contrast, MeDIP-seq neither introduces any mutations nor requires uracil-tolerant DNA polymerase. In terms of low amounts of starting DNA, MeDIP-seq requires as little as 1 ng of DNA, but this amount is insufficient for BS-seq because bisulfite conversion will destroy the majority of DNA. The convenient one-step immunoprecipitation in the MeDIP-seq procedure shows the merits of profiling genome-wide 5mC, 5hmC, 5-formylcytosine, and 5-carboxylcytosine in parallel with low amounts of starting DNA as long as the specificity of antibodies is verified (Shen et al., 2013). However, bisulfite conversion-based methods (Tet-assistant BS-seq and oxidative BS-seq) require additional treatments, such as labeling and oxidation, that will lead to further degradation and significant loss of DNA (Booth et al., 2012; Ficz et al., 2011; Yu et al., 2012). The double degradation by oxidation and bisulfite treatment (oxBS-Seq) makes it impossible for 1 ng of starting DNA to be sufficient because typically there will be less than 0.01 ng of DNA left after the oxBS procedure.
Recent BS-seq studies on genome-wide DNA methylation in oocytes and early embryos required several hundreds of blastocysts and thousands of oocytes, and they were not able to apply this technology to single germ cells (Kobayashi et al., 2012; Smallwood et al., 2011). In contrast, we only used 1 ng of DNA for MeDIP-seq, which lowers the starting DNA to approximately 100–200 cells, i.e., only a few blastocyst-stage embryos. We are not aware of any previously published MeDIP-seq studies beginning with such a low amount of DNA, although 1 ng of DNA could be applied to the RRBS method and tagmentation-based whole-genome bisulfite sequencing (Adey and Shendure, 2012; Schillebeeckx et al., 2013). The specialized nature of performing MeDIP-seq on low quantities of starting material and the difficulty in standardization may explain the lack of commercially available kits. Further optimization of this MeDIP-seq protocol may generate a DNA methylome from single cells.
Spike-in controls are usually employed for quality control of MeDIP-seq efficiency. Although the spike-in homologous fragments are different from genomic DNA, they can at least provide indirect information about immunoprecipitation efficiency (Taiwo et al., 2012). We added 10 pg of control DNA to 100 ng, 10 ng, and 1 ng of genomic DNA samples and found the immunoprecipitation efficiency to drop slightly with the decrease of starting DNA (Fig. 2B). This occurred because the absolute control of DNA loss is increased along with the decrease of companion genomic DNA from 100 ng to 1 ng in the presence of the same amount of antibodies. A similar trend for control immunoprecipitation efficiency decrease was also observed by using 5hmC antibody (Fig. 2B). In contrast, the endogenous methylation region appeared to be more enriched in low, rather than high, concentrations of starting DNA (Fig. 2C). The 5mC or 5hmC antibodies bind preferentially to highly methylated/hydroxymethylated fragments when low amounts (1 ng) of single-stranded DNA are present in the solution. The MeDIP procedure would fail to identify the hypomethylated (CpG poor) compared to hypermethylated (CpG rich) regions. We tried to evaluate the relative enrichment versus unmethylated Gapdh or β-actin promoters in 1 ng of starting DNA but failed. Rare amounts of Gapdh or β-actin fragments were pulled down after immunoprecipitation due to their hypomethylation; therefore, not enough DNA was enriched for the following real-time qPCRs.
Potential biases, such as copy number variations, GC content, and CpG density bias, are thought to exist in MeDIP-seq data (Laird, 2010). The ratio of genomic DNA versus 5mC/5hmC antibody and specificity of primary antibodies may also introduce variations among MeDIP-seq data in individual laboratories. In addition, whole-genome amplification mediated by universal and index primers during high-throughput sequencing library preparation may contribute to MeDIP enrichment bias, especially at certain CpG-rich targets (Borgel et al., 2012). Therefore, it is necessary to validate MeDIP-seq data by other DNA methylation detection approaches, such as bisulfite sequencing.
Because low amounts of genomic DNA (1 ng) are not enough for BS-seq, it is convenient to assess the MeDIP-seq data by examining commonly known methylated and unmethylated genomic loci. The CpG islands in housekeeping gene promoters are generally unmethylated (Jones, 2012). In mESCs, pluripotency genes are usually highly expressed and their promoters are hypomethylated. However, the lineage-specific gene promoters are methylated to prevent transcriptional initiation and elongation (Fouse et al., 2008; Imamura et al., 2006). In addition, the DMRs of imprinted control regions at imprinted genes are allele-specific methylated: One allele is methylated whereas the other is unmethylated to facilitate parent-of-origin expression (Bartolomei and Ferguson-Smith, 2011). In this study, a handful of selected methylated loci in gene bodies and promoters were relatively enriched in the MeDIP-seq data (Tables 3 and 4). The promoter regions of pluripotency genes and housekeeping genes are off-targets by the MeDIP-seq procedure because of their low methylation level. In contrast, upstream regions of H19 and Igf2r where the differentially methylated regions reside were more than three times enriched. Therefore, at least these commonly known genes were verified in our MeDIP-seq data by using 1 ng of DNA, further illustrating the reliability of the MeDIP-seq protocol.
Footnotes
Acknowledgments
We would like to thank Drs. Mingyi Zhou and Nathan Bivens at University of Missouri DNA core facility for MeDIP-seq library construction and Illumina deep sequencing, and acknowledge funding from Food for the 21st Century at the University of Missouri, and USDA-NRSP8.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.