
Abstract
Abstract
The essence of the reprogramming activity of somatic cell nuclear transfer (SCNT) embryos is to produce normal fertilized embryos. However, reprogramming of somatic cells is not as efficient as the reprogramming of sperm. In this report, we describe the effect of an inducible, specific miR-34 microRNA expression in donor cells that enables a similar level of sperm:transgene expression on the early development of SCNT embryos. Our results showed that donor cells with doxycycline (dox)-induced miR-34c expression for the preparation of SCNT embryos resulted in altered developmental rates, histone modification (H3K9ac and H3K4me3), and extent of apoptosis. The cleavage rate and blastocyst formation of the induced nuclear transfer (NT) group were significantly increased. The immunofluorescence signal of H3K9ac in embryos in the induced NT group significantly increased in two-cell- and eight-cell-stage embryos; that of H3K4me3 increased significantly in eight-cell-stage embryos. Although significant differences in staining signals of apoptosis were not detected between groups, lower apoptosis levels were observed in the induced NT group. In conclusion, miR-34c expression induced by dox treatment enhances the developmental potential of SCNT embryos, modifies the epigenetic status, and changes blastocyst quality.
Introduction
T
The current view on fertilization is that the main function of the sperm is to carry male DNA into egg cells and activate the fertilized oocytes. Numerous studies, however, have recently demonstrated that a wide range of RNA molecules, including messenger RNA (mRNA), transfer RNA (tRNA), small noncoding RNA, small interfering RNA (siRNA), and microRNA (miRNA), can be found in mature sperm (Amanai et al., 2006) and delivered into the oocyte during fertilization (Ostermeier et al., 2004). Some of these RNA molecules remain stable until embryonic genome expression is initiated, which indicates that some of them have an important role in zygotic gene activation and embryo development. The role of these sperm-derived RNA molecules, however, is incompletely understood.
miRNAs are a class of short (18–25-nucleotides) noncoding RNAs that bind to partially complementary sequences in mRNAs. These RNAs induce target mRNA translational silencing (incomplete match) and/or degradation (perfect complementarity), both of which result in downregulation of the protein encoded by the mRNA (Bartel, 2004). Some specific miRNAs have a key function reprogramming; miR-34 miRNA, for example, helps control embryonic stem cell (ESC) differentiation (Lin et al., 2005). However, miR-34c knockout mice enhance reprogramming by a lesser degree of repression of pluripotency genes, including Nanog, Sox2 and N-Myc (Choi et al., 2011). A recent study in mouse showed that miR-34c is an miRNA expressed specifically in sperm and necessary for the development of embryos, especially for cleavage of the fertilized embryos (Liu et al., 2012). Thus, we sought to determine whether or not the lack of miR-34c in donor cells is a key factor influencing the early development of bovine SCNT embryos. If yes, improving the expression of miR-34c in the donor cells to levels similar to that in sperm to enhance the development and reprogramming efficiency of SCNT embryos is a worthwhile research subject.
To address these issues, we adopted an inducible expression system of miR-34c used for donor cells; this system is called the Tet-On 3G System. Fan et al. (2012) used the Tet-On 3G response element promoter driving firefly luciferase to transduce blastocysts. Klymiuk et al. (2012) used the binary Tet-On System in combination with the SCNT technique to generate pig models with inducible transgene expression. This system, however, has been extensively tested in donor cells for nuclear transfer (NT) to prepare cloned embryos. The Tet-On 3G System is sensitive to doxycycline (dox) and precisely controls gene expression (Clontech, Mountain View, CA, USA). Tet-On 3G binds specifically to the TRE3G promoter and activates the transcription of the downstream precursor miR-34c only after dox is applied in the donor cell culture process (Fig. 1A). In the absence of dox, nearly nonexistent induced expression is observed because the TRE3G promoter lacks binding sites for endogenous mammalian transcription factors.
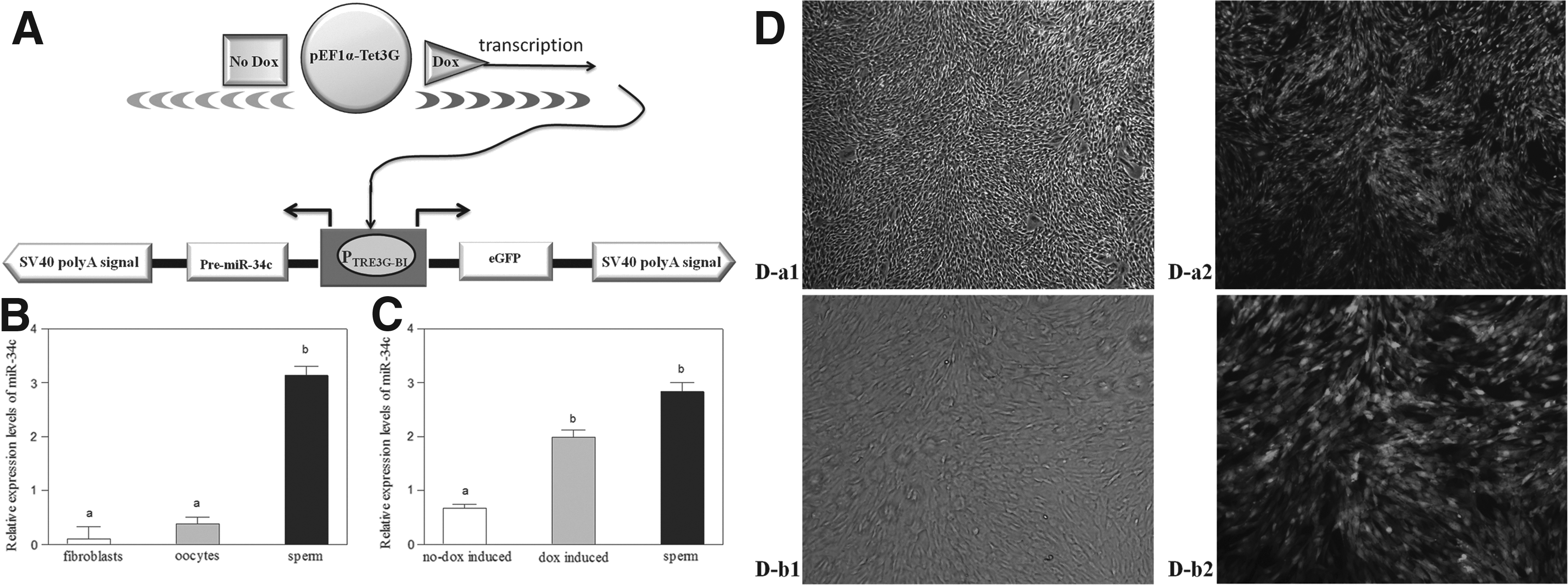
Preparation process and validation of donor cells. (
Successful nuclear reprogramming from a somatic state to an embryonic one is a key event in SCNT (Hochedlinger and Jaenisch, 2006). The nuclear reprogramming process mainly involves various epigenetic modifications wherein specific histone modifications are removed from the chromatin template and new marks are placed onto different regions of chromatin to facilitate expression of the embryonic genome (Li, 2002). Hence, we used the immunostaining method to examine the acetylation levels of histone H3K9 (H3K9ac) and trimethylation levels of H3K4 (H3K4me3). We further assessed the quality of embryos using the terminal dexynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay. In this work, the following groups were tested: In vitro–fertilized embryos (IVF group), dox-induced/treated SCNT embryos (induced NT group), and no dox-induced/treated SCNT embryos (control NT group).
Materials and Methods
Construction and validation of the Tet-On 3G vector
The pre-miR-34c sequence (bovine precursor microRNA-34c) derived from miRBase completed gene synthesis and was connected to the KpnI–PstI sites of the PUC57 vector produced by GenScript Co. (Nanjing, China). Double enzyme digestion and recycling yielded the pre-miR-34c fragment, which was inserted into the MCS-2 site of the pTRE3G-BI vector. MCS-1 on the right side of pTRE3G-BI was replaced with the enhanced green fluorescent protein (eGFP) sequence (BamHI—NotI sites) derived from pd1EGFP-N1 (Clontech, Mountain View, CA, USA) (Fig. 1A). High-quality plasmid DNA was isolated (PureYield Plasmid Midiprep, Promega, Madison, WI, USA) for cell transfection, and a small portion of plasmid was used for sequencing by GenScript Co.
Cell culture, transfection, and selection
Primary fibroblast cell cultures were established from approximately 35-day-old fetuses, as described previously (Liu et al., 2013). Briefly, after the head and viscera were removed, the remaining tissue was rinsed several times with phosphate-buffered saline (PBS) and minced into 1 mm3 pieces. The tissue pieces were cultivated for 1–2 weeks in 60-mm Petri dishes with Dulbecco's Modified Eagle's Medium (DMEM; Gibco, New York, NY, USA) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 100 U of penicillin, and 250 ng of amphotericin B (Gibco) at 37°C in 5% CO2. When fibroblast cells from the tissue pieces had reached 90% confluency, they were treated with 0.5 mL of TE buffer (0.25% trypsin/0.05% EDTA) and passaged 1:2 or 1:3. The cells were then trypsinized and frozen in 50% FBS, 40% medium, and 10% dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) for long-term storage and future use. When needed, cells were thawed and grown in DMEM/F12 (Gibco) medium supplemented with 15% FBS and incubated at 37°C in a 5% CO2 atmosphere. At 70–80% confluency, cells were harvested by trypsinization and resuspended in 2 mL of Opti-MEM (Gibco).
The cells were placed in a 4-mm cuvette gap with 10 μg of the pTRE3G-BI response plasmid (Amp-pre-miR-34c-pTRE3G-BI-eGFP) and 5 μg of the pEF1α-Tet3G regulator plasmid and then electroporated at 510 V with three pulses of 1-msec duration using a BTX Electro Cell Manipulator ECM2001 (BTX Harvard Apparatus, San Diego, CA, USA). Electroporated cells were mixed with fresh cell culture medium and placed on 90-mm Petri dishes at an average density of 5×105 cells. After 48 h, the culture medium in the Petri dishes containing 700 mg/mL G418 (Sigma) was changed and replaced once every 3 days. Approximately 7 days later, G418-resistant colonies began to emerge and dox was added at a dosage of 1000 ng/mL. When the colonies had become large enough to transfer, we used a marker pen to indicate fluorescent colonies, harvested large, healthy colonies, and transferred each colony into separate wells of a 48-well plate. The colonies were cultured in 500 μg/mL G418. When confluent, cells from each well were split into three wells of a 48-well plate for further validation of the expression of nuclear donor cells.
Oocyte collection and in vitro maturation
Oocyte collection and in vitro maturation (IVM) were performed using standard techniques (Wang et al., 2011). Briefly, cumulus–oocyte complexes (COCs) were obtained by aspiration (a 12-gauge needle attached to a 10-mL syringe) of surface-visible follicles (diameters between 2 and 8 mm) from the ovaries of slaughtered bovine. After the COCs had been washed twice in PBS containing 5% (vol/vol) FBS, oocytes surrounded by a minimum of three cumulus cell layers and with uniform cytoplasm were selected. The COCs were the cultured for 20 h in bicarbonate-buffered Tissue Culture Medium-199 (TCM-199, Gibco) containing 10% (vol/vol) FBS, 1 mg/mL 17β-estradiol, and 0.075 IU/mL human menopausal gonadotropin in 95% humidified air with 5% CO2 at 38.5°C.
Preparation, activation, and culture of SCNT embryos
SCNT, activation of reconstructed embryos, and culture of SCNT embryos were performed as described previously (Wang et al., 2011). Briefly, after IVM for 20 h and dispersal of the COCs by vortexing of the cumulus cells in PBS supplemented with 0.1% bovine testicular hyaluronidase, oocytes with the first polar body (metaphase II) and evenly granulated ooplasm were selected for enucleation. The first polar body and a small amount of the surrounding cytoplasm were removed from the oocytes with a glass pipette (inner diameter, 20 mm) using PBS microdrops supplemented with 7.5 mg/mL cytochalasin B and 10% FBS. Two groups of cells (with dox and without dox) were used as donor cells. A single donor cell was placed in the perivitelline space of the enucleated oocytes. The oocyte–cell couplet was sandwiched between a pair of platinum electrodes connected to a micromanipulator with microdrops of Zimmermann's fusion medium. A double electrical pulse of 35 V for 10 μsec was applied for oocyte–cell fusion. Reconstructed embryos were kept in modified synthetic oviductal fluid (mSOF) (Wang et al., 2012) containing 5 mg/mL cytochalasin B for 2 h until activation. Activation of reconstructed embryos was performed in 5 μM ionomycin for 4 min, followed by 4 h of exposure to 1.9 mM dimethylaminopyridine in mSOF.
After activation, embryos were cultured in G1.5/G2.5 (Vitrolife AB, Gothenburg, Sweden) sequential medium. Droplets of 150 mL of G1.5 were prepared in a 35-mm cell culture dish under mineral oil (20–30 embryos per microdrop). Embryos were transferred to G2.5 droplets on day 3 of culture (day 0 being the day SCNT was performed).
In vitro fertilization
IVF was applied based on standard methods (Wang et al., 2012) with some modifications. Briefly, frozen thawed semen was placed at the bottom of a 15-mL tube with 5 mL of Brackett and Oliphant (BO) medium supplemented with 6 mg/mL bovine serum albumin (BSA) and 20 mg/mL heparin and then incubated for 30 min at 38.5°C in a humidified atmosphere of 5% CO2. The top 3.5 mL of the BO medium was removed and centrifuged at 1000×g for 10 min. A 50-μL sperm suspension (concentration, 2×106 spermatozoa/mL) was added to 35–40 COCs in a 50- to 80-μL microdrop of the BO medium under mineral oil.
After microdrop IVF for 20 h, oocytes were washed with SOF to disperse cumulus cells and redundant spermatozoa. The oocytes were then washed again and cultured in droplets of 400 μL of G1 medium (Vitrolife AB) under mineral oil (0 h being the time embryos were transferred into the G1 medium).
Quantitative real-time PCR
Three blastocysts, five oocytes at the metaphase II stage, and five embryos in the same batch were randomly chosen and pooled for total RNA extraction. Samples were lysed using resuspension buffer and RNaseOUT™ from a SuperScript™ III CellsDirect cDNA Synthesis System (Invitrogen, Carlsbad, CA, USA) based on the manufacturer's protocol. Fibroblast cells (donor cells) and spermatozoa were prepared by squeezing. Lysing of the cauda epididymis was conducted using TRIzol reagent (Invitrogen) based on the manufacturer's instructions.
The reverse transcription reaction was achieved using a miScript II RT Kit (Qiagen, Hilden, Germany) based on the manufacturer's procedure. Mature miRNAs were polyadenylated by poly(A) polymerase and converted into cDNA by reverse transcriptase. miR-34c was quantified using SYBR Premix Ex Taq™ II (TaKaRa, Otsu, Japan) on a StepOne real-time polymerase chain reaction (PCR) system (StepOne and StepOnePlus; ABI, Foster City, CA, USA) based on the manufacturer's protocol. U6 small nuclear RNA was used as an internal control.
Immunofluorescence staining of embryos
Immunofluorescence staining was conducted in accordance with the methods of our previous study (Su et al., 2011). Briefly, the collected embryos were fixed with Immunol Staining Fix Solution (Beyotime, Jiangsu, China) for 30 min and then permeabilized for 30 min with 0.2% Triton X-100 in PBS–polyvinyl alcohol (PVA). After three washings, all samples were incubated overnight in Immunol Staining Blocking Solution (Beyotime) at 4°C and then incubated with the first antibodies to H3K9 acetylation (1:1,000; Abcam, Cambridge, UK) and H3K4 trimethylation (1:1,000; Abcam, Cambridge, UK) for 12 h at 4°C.
After three washings, secondary antibodies [Alexa Fluor 488-labeled goat anti-rabbit immunoglobulin G (IgG; 1:500; Beyotime) for H3K9ac and Alexa Fluor 488-labeled goat anti-mouse IgG (1:500; Beyotime) for H3K4me3 were incubated with the samples for 2 h. Finally, the DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime) for 3–5 min. All samples were mounted on glass slides with a drop of Antifade Mounting Medium (Beyotime) and analyzed using a Nikon Eclipse Ti-S microscope equipped with a 198 Nikon DS-Ri1 digital camera (Nikon, Tokyo, Japan).
Apoptosis assays
Apoptosis was determined using a DeadEnd Fluorometric TUNEL System (Promega) in accordance with the methods of our previous study (Su et al., 2011). Briefly, embryos were washed, fixed, and permeabilized for immunofluorescence staining. After equilibration in equilibration buffer, embryos were incubated with rTdT incubation buffer in the dark for 1 h at 37°C. The tailing reaction was terminated in 2× standard saline citrate for 15 min. Finally, the DNA was stained with DAPI (Beyotime). Samples were mounted on glass slides for immunofluorescence staining.
Experimental design and statistical analysis
Experiment 1
This experiment was examination of the expression level of miR-34c in sperm, oocytes (metaphase II), and bovine fetal fibroblasts by qRT-PCR (Table 1).
The miScript Universal Primer for detection of mature microRNA (Qiagen).
F, forward; R, reverse.
Experiment 2
Here nuclear donor cells expressing miR-34c were prepared, and the Tet-On 3G vector was constructed and validated using standard techniques. pEF1α-Tet3G and pTRE3G-BI plasmids were transfected into fibroblast cells using the BTX Electro Cell Manipulator ECM2001. Select cells were cultured with 700 mg/mL G418. The same colonies were divided into two groups, namely, the dox-induced/treated and no dox-induced/treated groups.
Experiment 3
In this experiment, SCNT embryos were evaluated using nuclear donor cells expressing miR-34c. The cleavage rates (days 2 and 7) of IVF (IVF group) and SCNT (induced NT group and control NT group) embryos were recorded to assess the in vitro developmental capacity of each group. Cleavage rates were determined at the two-cell, four-cell, eight-cell, and blastocyst stages to detect H3K9ac and H3K4me3 levels.
The 2−ΔΔT method (Livak and Schmittgen, 2001) was used to quantify the relative mRNA levels. Using Image-Pro Plus for analyzing fluorescence intensity, as described previously (Das et al., 2010), all individual nuclei of embryos were outlined. Then, integrated optical density (IOD) and area were measured after the calibration of OD. The average normalized fluorescence intensity for a single embryo was represented by “sum IOD/sum area.” Finally, H3K9ac and H3K4me3 levels were divided by the total DNA contents (DAPI total fluorescence intensities) to calculate normalized AcH3K9 and H3K4me3 quantities, respectively. To minimize the difference among embryos, all images were obtained with the same exposure times and adjustments of the microscope. The experiments were replicated three times with five to 10 embryos per group. The level of histone acetylation or histone methylation of embryos was presented as the mean value of embryos±standard error of the mean (SEM). To quantify fluorescence intensity, the intensity levels of the induced NT group and control NT group embryos were presented relative to the mean intensity level of IVF embryos.
The expression levels of miR-34c and fluorescence intensity were analyzed by one-way analysis of varianc (ANOVA) and Tukey's least significant difference test using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered significant at p<0.05. Continuous data are presented as mean±SEM.
Results
The relative mRNA expression of miR-34c in oocytes (metaphase II), sperm, and bovine fetal fibroblasts is presented in Figure 1B. Expression levels of miR-34c were significantly higher in sperm than in oocytes (metaphase II) or bovine fetal fibroblasts (p<0.05).
Construction of the Tet-On 3G vector was completed by inserting the pre-miR-34c fragment (KpnI–PstI sites) into MCS-2 and the eGFP sequence (BamHI–NotI sites) into MCS-1 of the pTRE3G-BI vector (Fig. 1A).
After the pEF1α-Tet3G and pTRE3G-BI plasmids had been collectively transfected into fibroblast cells, colonies were selected for culture with G418. When only dox was added to the culture medium, colonies of bovine fetal fibroblasts showed fluorescence (Fig. 1D-a2 and 1D-b2). In the absence of dox, nearly nonexistent fluorescence is observed. Not all of the selected colonies survived isolation and expansion. Thus, colonies were strictly screened for high inducibility by green fluorescence after dox addition (Fig. 1D) because only two types of plasmids may be transfected into fibroblast cells. We then harvested dox-induced and no dox-induced fluorescent colonies and compared the relative abundances of miR-34c in these colonies with that of sperm (Fig. 1C). miR-34c expression was significantly higher in sperm and dox-induced fluorescent colonies than in no dox-induced fluorescent colonies (p<0.05). miR-34c expression in dox-induced fluorescent colonies was not significantly different from that in sperm.
Developmental rates of the cleavage and blastocyst stages of the SCNT embryos were analyzed from four replicates (Fig. 2; Table 2). IVF and all SCNT embryos cleaved showed similar rates, ranging from approximately 72% to 77%. The rates of the induced NT and IVF groups were significantly higher than that of the control NT group (p<0.05). The rates of blastocyst stages of the induced NT and IVF groups were also significantly higher than that of the control NT group (p<0.05). No significant difference in embryo developmental rates between the induced NT and IVF groups was observed.
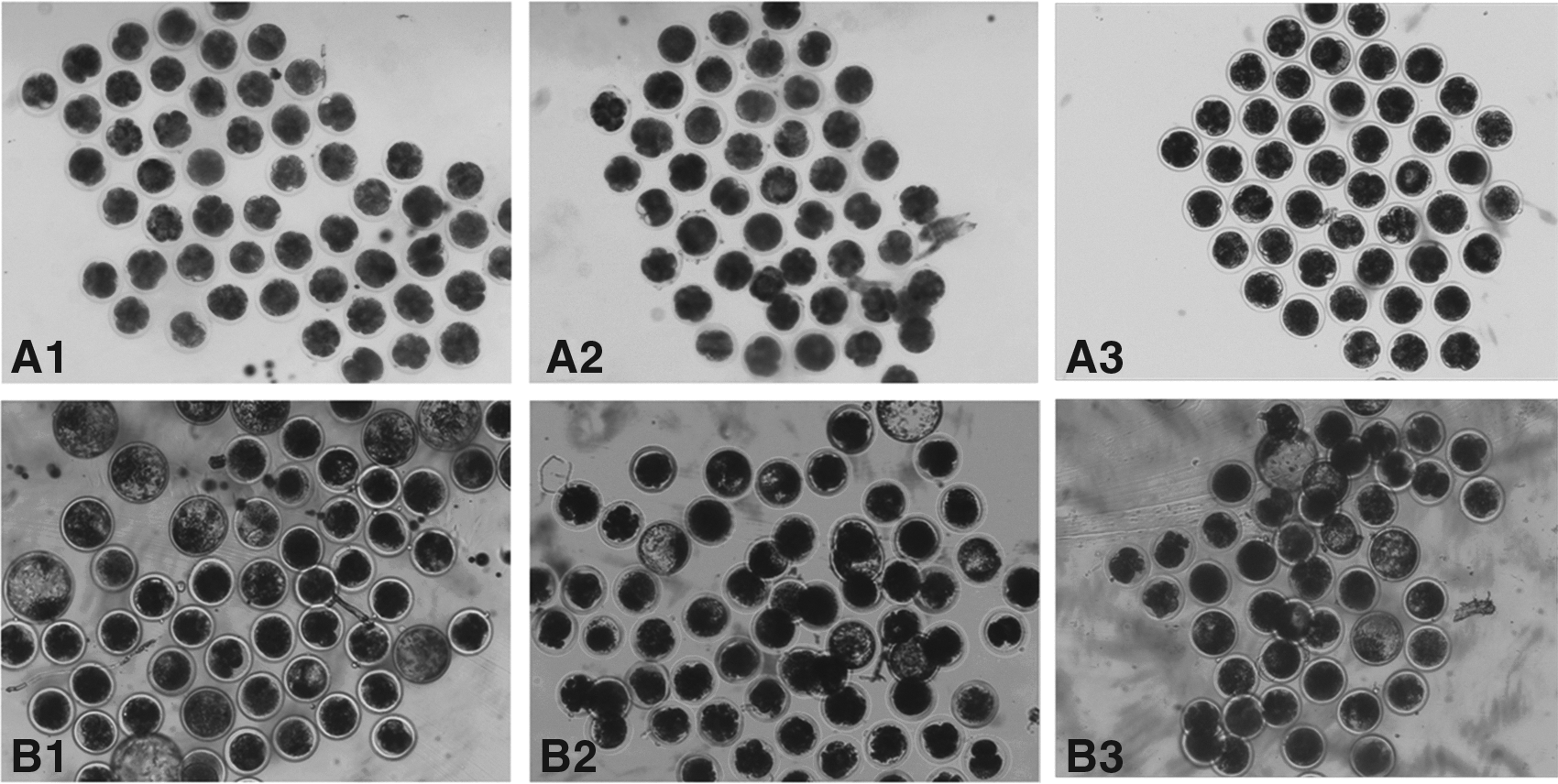
Representative photographs of bovine NT embryo cleavage (magnification, 40×) on day 2 (
Four replicates were performed. Numbers in parentheses represent development rates [mean±percent change in standard error of the mean (SEM%)], whereas other numbers represent total embryo numbers of four replicates.
Values with different superscript within columns are significantly different from each other (p<0.05).
SCNT, somatic cell nuclear transfer; NT, nuclear transfer; IVF, in vitro fertilization.
The acetylation levels of histone H3K9 and trimethylation levels of H3K4 were examined in two-cell stage embryos, eight-cell stage embryos, and blastocysts by the immunostaining method (Fig. 3A). No confusing signals were detected in embryos stained without first or secondary antibodies. The fluorescence intensity of H3K9ac and H3K4me3 was measured in two-cell-stage embryos, eight-cell-stage embryos, and blastocysts (Fig. 3B). As shown in Figure 3, the relative fluorescence intensity of H3K9ac in two-cell-stage and eight-cell-stage embryos of the induced NT group was significantly higher than that obtained from embryos of the control NT group (p<0.05). The relative fluorescence intensity of H3K9me3 in eight-cell-stage embryos of the induced NT group was significantly higher than that observed in embryos of the control NT group (p<0.05). No differences in H3K9ac and H3K9me3 levels at the blastocyst stage were observed among groups. Differences in staining signals of H3K4me3 were also undetected in two-cell-stage embryos.
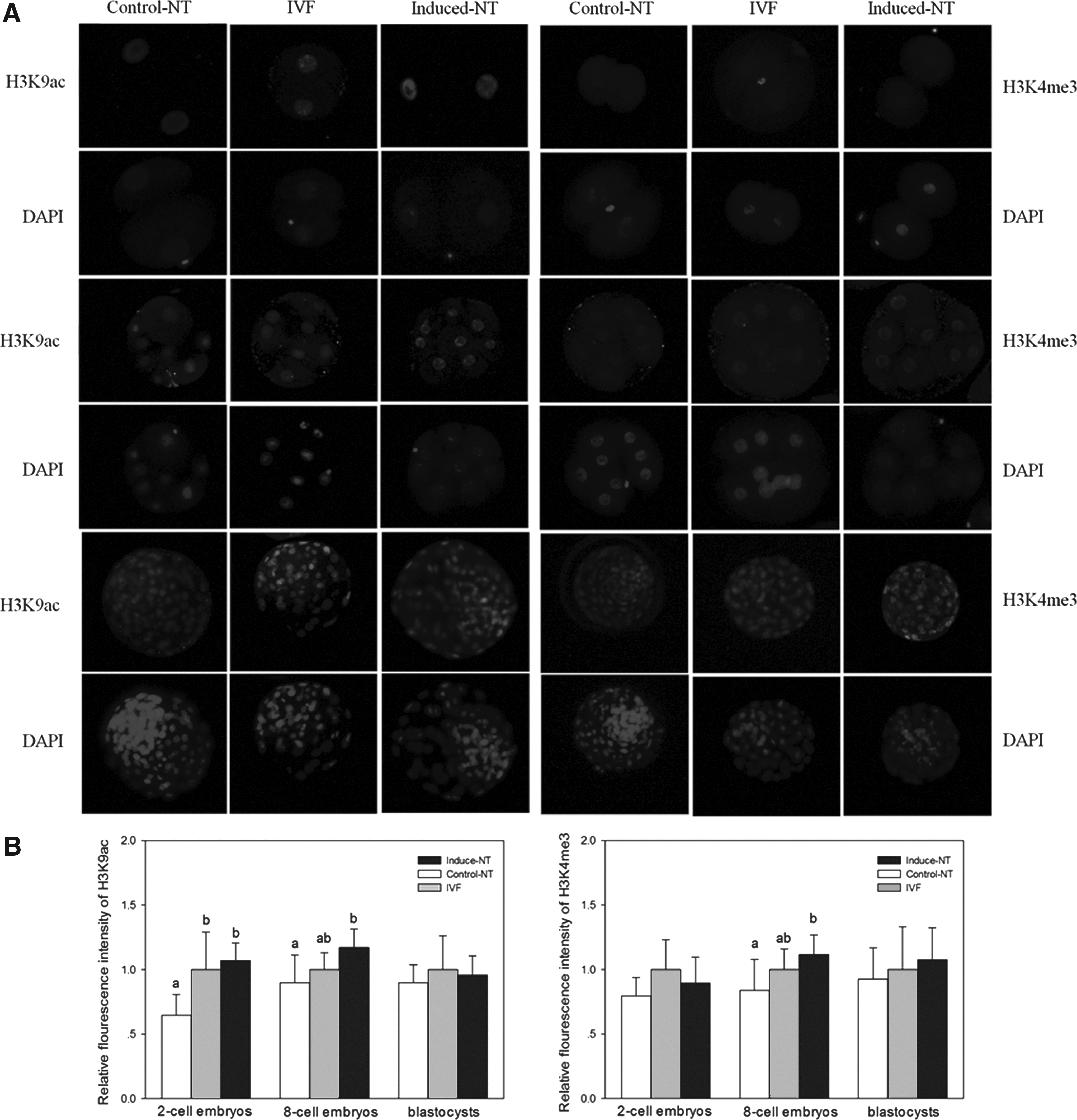
(
The total number of apoptotic cells was determined by observing fluorescein isothiocyanate (FITC)-conjugated 2′-deoxyuridine-5′-triphosphate-labeled nuclei and comparing DAPI-counterstained nuclei. TUNEL assay results are shown in Figure 4. The number of apoptotic cells in IVF blastocysts was significantly lower than that in cells in the control NT group (p<0.05) (Fig. 4B). No significant differences between IVF blastocysts and induced NT blastocysts were observed. As well, the extent of apoptosis in the induced NT group was lower than that in the control NT group; differences observed, however, were insignificant.
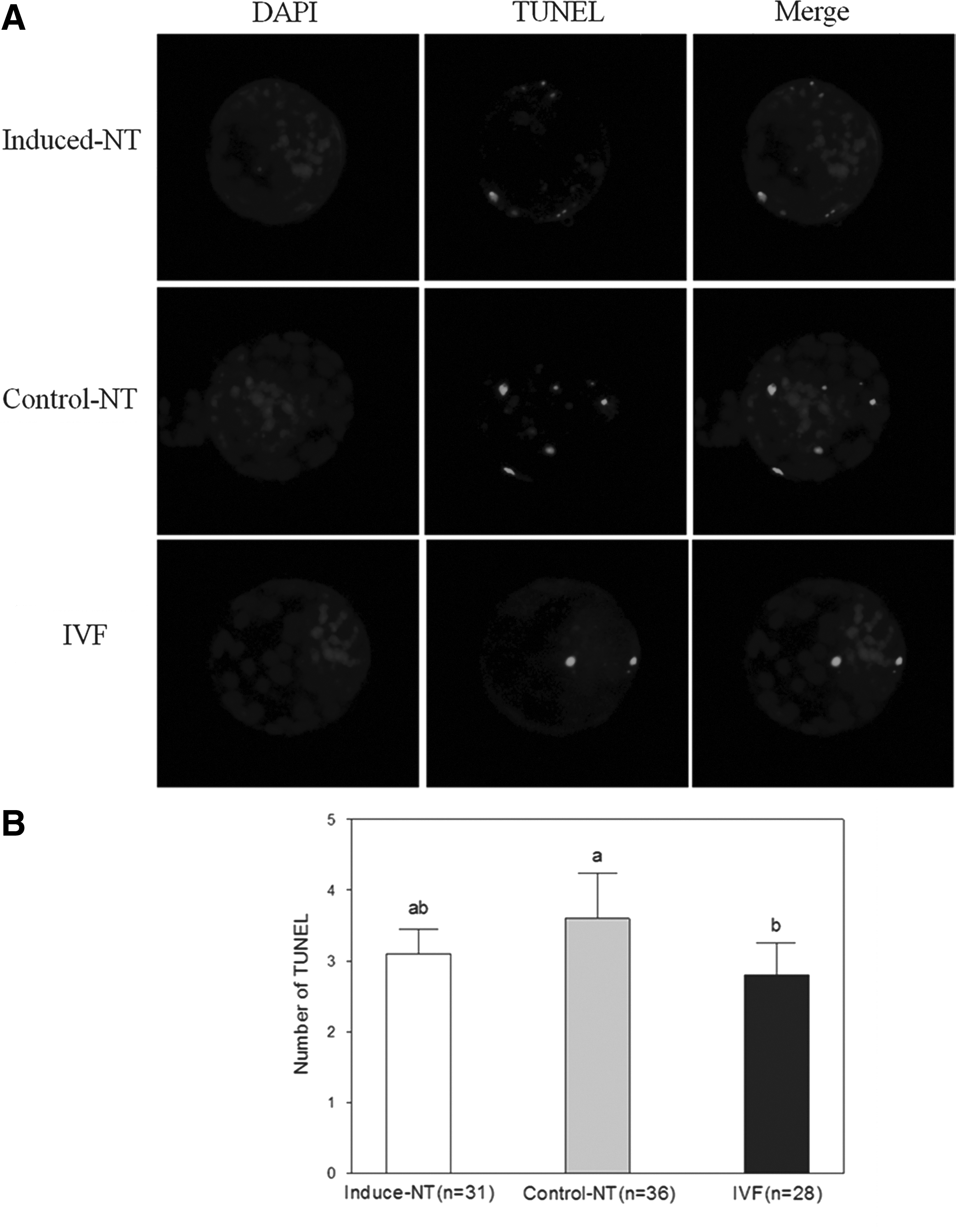
Apoptosis in blastocysts. (
Discussion
Recent studies have shown that sperm carries numerous RNAs into oocytes during fertilization (Miller and Ostermeier, 2006; Ostermeier et al., 2004). RNA molecules, which are also considered key vectors of epigenetic variation, are obviously the best candidates for guiding the functions of protein complexes and other components of silencing/activating systems by sequence recognition. These molecules could serve as templates for the synthesis of transcription factors important for embryogenesis (Kumar et al., 1993) and directly regulate embryonic development. Indeed, sperm-borne miR-34c is required for first-cleavage division in mouse (Liu et al., 2012). Collective evidence indicates that sperm RNAs may have an important function in the development of fertilized embryos. Such observations present new ideas that may improve the SCNT technique arouse a particular interest. In the present study, the effects of miR-34c, a microRNA expressed mainly in sperm, on the development of bovine SCNT embryos were assessed.
Evidence indicates that miR-34c is highly expressed only in germ cells of spermatogenesis (Bouhallier et al., 2010). In this study, we proved that miR-34c of mRNA expression level is significantly higher in sperm than that in metaphase II oocytes and bovine fetal fibroblasts (p<0.05) (Fig. 1B). Although virus gene delivery and simple plasmid vector systems have been widely used for achieving transgenesis in cattle (Gong et al., 2004; Hofmann et al., 2004), we considered the particularity of donor cells in avoiding constant expression of the miR-34c. To conform to expression patterns of the normal fertilized zygote, we used the Tet-On 3G gene delivery system in this study for donor cells of bovine fetal fibroblasts (Fig. 1A). This delivery system has been successfully applied in reversible and dose-dependent induction of expression (Gossen et al., 1995). After addition of dox to the cultures of G418-resistant colonies, Tet-On 3G bound specifically to the TRE3G promoter and activated transcription of the downstream precursor miR-34c (Fig. 1A). As such, we ensured that the pEF1α-Tet3G and pTRE3G-BI plasmids were transferred into the fibroblast cells at the same time.
We then added different concentrations of dox to determine high induction and low basal expression to ensure that the expression level of miR-34c in fibroblast cells approaches that in sperm. miR-34c expression was significantly higher in sperm and dox-induced fluorescent colonies than in no dox-induced fluorescent colonies (p<0.05). miR-34c expression in dox-induced fluorescent colonies was not significantly different from that in sperm (Fig. 1C).
In a recent study in mice, over 70% of the zygotes injected with the miR-34c inhibitor failed to cleave (Liu et al., 2012). After completion of the preparation of SCNT embryos, we investigated whether or not dox-induced expression of miR-34c influences the developmental capacity of bovine SCNT embryos in vitro. We determined that the developmental rates of IVF and all SCNT embryos cleaved were similar (Table 2). However, the cleavage rates and blastocyst stages of the induced NT group were significantly higher than those of the control NT group (p<0.05). These results indicate that miR-34c improves the in vitro development of SCNT bovine embryos.
We examined and compared the acetylation levels of histone H3K9 and trimethylation levels of H3K4 in two-cell-stage embryos, eight-cell-stage embryos, and blastocysts (Fig. 3A). Histone acetylation has a significant function in the process of reprogramming and affects the development of SCNT embryos (Dai et al., 2010; Das et al., 2010). We determined that induction of miR-34c expression promotes increased H3K9ac levels in two-cell and eight-cell SCNT embryos. Trichostatin A, an effective histone deacetylase (HDAC) inhibitor, is often used to increase levels of histone acetylation and can improve the full-term development of SCNT embryos (Iager et al., 2008). Sirtuin 1 (SIRT1), a member of the HDAC family, can be regulated by miR-34a (Yamakuchi et al., 2008). miR-34c may analogously regulate the targeting of SIRT1 mRNA and/or other targets to affect histone acetylation. Other results show increased H3K4me3 levels in SCNT embryos at the eight-cell stage. Bovine embryonic development into eight cells is extensively recognized to occur concomitantly with the activation of the embryonic genome.
H3K4me3, a marker associated with active transcription, is also important in the efficient reprogramming of pluripotency genes (Eissenberg and Shilatifard, 2010; Murata et al., 2010). Several factors have been reported to associate directly with H3K4me3, including the CHD1 protein and proteins containing a plant homeodomain finger domain, such as ING2, ING4, ING5, RAG2, and BPTF (Pray-Grant et al., 2005; Shi et al., 2006; Shi et al., 2007; Sims et al., 2005; Wysocka et al., 2006). We suggest that increased H3K4me3 expression may activate transcription of these genes and alter efficient reprogramming. No differences in H3K9ac and H3K9me3 levels at the blastocyst stage were detected among groups. Although early SCNT embryos showed various incorrect histone modifications, the final blastocysts showed similar histone modification. Abnormal histone modification at early embryonic stages may be corrected at the blastocyst stage (Wu et al., 2011).
Apoptosis can eliminate cells with nuclear or chromosomal abnormalities and helps evaluate the quality of blastocyst (Hardy, 1997). SCNT blastocysts with high apoptosis levels show a diminution in total cell number (Yu et al., 2007). In the present study, we observed no significant differences in number of apoptotic blastocysts between the induced NT and control NT groups (Fig. 4). miR-34–induced apoptosis presumably depends on the cellular context and, therefore, the expression levels of the respective miR-34 target proteins involved in the regulation of apoptosis (Hermeking, 2010). Evidence indicates that the action of miR-34c may depend on the balance of specific intracellular mediators of apoptosis (Catuogno et al., 2013). Hence, we observed no significant differences in the interplay between miR-34c, p53, and target genes between the two groups. Although evidence indicates that suppression of reprogramming by miR-34c, as a target p53 signal, was due to mild repression of pluripotency genes, including Nanog, Sox2, and N-Myc (Choi et al., 2011), the final blastocysts in the present study show the histone modification similarly (Fig. 3). This study that imposing sperm-like states on nuclear donor cells by dox treatment will make them more prone to reprogramming at fertilization, but zygotes in subsequent developmental stage are not treated by dox. Specific mechanisms need further research.
In summary, the results suggest that miRNAs are essential components in early embryonic development. Inducible miR-34c expression influences the early development of bovine SCNT embryos and may not only increase the cleavage rates of developing embryos but also change the quality of the resulting SCNT embryos. Further investigation is necessary to identify which genes are regulated by miR-34c.
Footnotes
Acknowledgments
The authors thank Mr. Younan Wang for transportation of the cow ovaries used in this study. This work was supported by a grant from the National High Technology Research and Development Program (#2011AA100303).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.