
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) are promising candidates for the study of disease models as well as for tissue engineering purposes. Part of a strategy to develop safe reprogramming technique is reducing the number of exogenous reprogramming factors. Some cells types are more prone to reprogramming than others. iPSC induction with less reprogramming factors has been described in cells with endogenous expression levels of pluripotency genes, such as neural stem cells. Because multipotent neural crest stem cells (NCSCs) from mammalian hair follicle bulges also express pluripotency genes, we argued that this property would facilitate reprogramming of hair follicle bulge NCSCs and could substitute for the use of exogenous reprogramming factors. Although we confirmed the expression of pluripotency genes in hair follicle bulge cells, our results show that these cells do require a full set of reprogramming factors for iPSC induction. Hair follicle bulge–derived iPSCs were created with efficiencies similar to fibroblasts. We conclude that high endogenous levels of pluripotency factors are no guarantee for facilitated induction of pluripotency.
Introduction
M
The neural crest is a population of migratory multipotent progenitor cells in vertebrates that emerges and delaminates from the dorsal neural tube during neurulation. It contributes significantly to the development of mammalian craniofacial tissues (for review, see Santagati and RijLi, 2003). As a result, neural crest stem cells (NCSCs) persist in several craniofacial structures in the adult organism and therefore are a relatively accessible source of stem cells for transplantation studies and tissue engineering (for review, see Kaltschmidt et al. 2012; Shakhova and Sommer, 2010).
We isolated nestin-expressing NCSCs from the adult mouse whisker follicle (Amoh et al. 2005; Amoh et al., 2009; Sieber‐Blum et al. 2004; Sieber-Blum and Hu, 2008) and analyzed the expression of pluripotency genes in these adult hair follicle NCSCs (HFNCSCs) both at the mRNA and protein levels. We hypothesized that (high) endogenous expression of one or more pluripotency factors might facilitate pluripotency induction compared to, for instance, skin fibroblasts and/or that less transcription factors would be necessary to induce pluripotency. We confirmed the pluripotent expression profile of hair follicle bulge NCSCs and also showed that this does not facilitate their reprogramming into iPSCs and that a full set of Yamanaka factors is still required.
Material and methods
Isolation of HFNCSCs and cell culture
NCSCs were isolated from mouse whisker follicles as previously described (Sieber-Blum et al., 2004). In short, whiskers of C57BL/6 mice and nestin–green fluorescent protein (GFP) transgenic mice were dissected. Hair follicle bulges were cultured on poly-
iPSC induction
For reprogramming of the mouse HFNCSCs, we have used both the retroviral and lentiviral transduction approaches. For retroviral transduction, retroviral particle–containing supernatants were produced using Phoenix Eco packaging cells that were transfected using FuGENE, with separate pMX-based vectors encoding murine Oct4, Sox2, Klf4, and cMyc plasmids (Addgene), as described previously (Czepiel et al., 2011). For lentiviral transduction, HEK293T cells were transfected with a lentiviral self-inactivating pRRL.PPT.SF plasmid containing murine Oct4, Klf4, Sox2, c-Myc, and dTomato complementary DNAs and co-transfected with pMD2-VSV-G, and pCMV-Δ8.91 packaging vectors. Lentiviral supernatant was collected 48 h posttransfection and filtered through a 0.45-μm filter (Whatman). Around 20,000 HFNCSCs were transduced with viral supernatant supplemented with 8 μg/mL Polybrene (Sigma-Aldrich), which was replaced by fibroblast medium after 24 h. Transfection efficiency was evaluated on the basis of fluorescent dTomato expression. Four days posttransduction, cells were dissociated using 0.05% trypsin-EDTA and seeded onto irradiated mouse embryonic fibroblast (MEF) feeder layers on a 1% gelatin-coated six-well plate and cultured in ES medium. Colonies were counted after 7 days, and four clones were isolated using a binocular microscope and subcultured by passaging with trypsin-EDTA and culturing on irradiated MEFs (iMEFs) in ES medium. Previously generated fibroblast-derived iPSCs (F-iPSCs) and IB10 ESCs were also cultured on iMEFs in ES medium.
Embryoid body formation
For embryoid body (EB) formation, HFNCSC-derived iPScs (HF-iPSCs) were dissociated using 0.25% trypsin/EDTA and grown for 8 days as suspension cultures in 10-cm low-attachment petri dishes in EB medium consisting of knockout Dulbecco's Modified Eagle Medium (KO-DMEM) supplemented with 15% fetal calf serum (FCS), 2 mM
Immunocytochemistry
Immunocytochemistry was performed as previously described (Czepiel et al., 2011). The following antibodies were used: Mouse anti-mouse nestin (1:200; Chemicon, MAB 353), mouse anti-mouse Oct4 (1:100; Santa Cruz, 5279), rabbit anti-mouse Sox2 (1:200; Abcam, 15830), mouse anti-mouse Sox2 (1:200; Cell Signaling, 4900S), rabbit anti-Nanog (1:200; Abcam, 80892), mouse anti-mouse SSEA-1 IgM (1:200; Santa Cruz, 21702), mouse anti-mouse desmin (1:200; Dako M0760), mouse anti-mouse GATA (1:200; Santa Cruz, 25310), and mouse anti-mouse βIII-tubulin (1:400; Abcam 7751). The following fluorescently labeled secondary antibodies were used: Alexa Fluor 488-conjugated donkey anti-rabbit (1:300; Molecular Probes, A21206), Alexa Fluor 488-conjugated goat anti-mouse IgM (1:300; Invitrogen, M31601), DyLight 488-conjugated goat anti-rabbit (1:100; Jackson Immunoresearch), and Cy3-conjugated donkey anti-goat (1:300; Jackson Immunoresearch). For nuclear visualization, cells were counterstained with Hoechst 33342 (1:1000; Jackson Immunoresearch) in phosphate-buffered saline (PBS). The alkaline phophatase (AP) activity of iPSC colonies was detected using an AP detection kit (Sigma).
qRT-PCR
Total RNA was extracted (Chomczynski and Sacchi, 1987) and transcribed into cDNA using random hexamers and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Fermentas). Purity of the RNA was confirmed by means of gel electrophoresis. Quantitative real-time PCR was performed in 384-well plates using iQ SYBR Green Supermix (Bio-Rad) on an AB17900HT system (Applied Biosystems) with the following cycling parameters: 95°C for 3 min, 39 cycles with 95°C for 10 sec, and 58°C for 30 sec. qRT-PCR primers were designed with PerlPrimer for the transcripts listed in Table 1. Housekeeping genes hydroxymethylbilane synthase (HMBS) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as internal controls for normalization. The data were processed with SDS version 2.3 analysis software (Applied Biosystems). The relative expression ratio of target genes was quantified using the 2−ΔΔCt calculation method (Schmittgen and Livak, 2008).
Western blot
Proteins were extracted from cell samples using lysis buffer supplemented with protease inhibitors. Cell lysates were sonicated and quantitated by DC Protein Assay (Bio-Rad, Hercules, CA), and proteins were separated on 15% sodium dodecyl sulfate polyacrylamide gels and then transferred onto polyvinylidene fluoride membranes. Membranes were incubated in Odyssey blocking buffer and incubated overnight in primary antibodies diluted in PBS+0.1% Tween-20. The following primary antibodies were used: Mouse anti-mouse Oct4 (1:1,000; Santa Cruz, 5279), mouse anti-mouse Sox2 (1:1,000; Cell Signaling, 4900S), rabbit anti-mouse Klf4 (1:1000; Abcam 34814), rabbit anti-mouse Nanog (1:1000; Abcam, 80892), mouse anti-mouse β-actin (1:10,000; Abcam 6276), and rabbit anti-mouse β-actin (1:10,000; AbD Serotec AHP1387). The following day, membranes were washed and incubated for 1 h in the fluorescence-conjugated secondary antibodies donkey anti-mouse IRDye 680CW (1:10,000; LI-COR Biosciences) and donkey anti-rabbit IRDye 800CW (1:10,000; LI-COR Biosciences). Blots were washed, scanned with the LI-COR Odyssey infrared imaging system (LI-COR Biosciences), and analyzed with Odyssey 2.0 software.
Results
Expression of pluripotency factors in HFNCSCs
We isolated HFNCSCs from the adult mouse whisker follicle of nestin–GFP transgenic mice as well as from C57Bl/6 mice (Fig. 1A). Explants were placed in laminin-coated six-well plates, and NCSCs were allowed to migrate out of the follicle bulges (Fig. 1B, C). For assessment of pluripotency gene expression with immunohistochemistry, C57BL/6 cells were replated in small badges of 10–100 cells on laminin and allowed to expand clonally in selective proliferation medium. In this way, pure populations of HFNCSCs could be obtained that proliferated in monolayers as described previously (Sieber-Blum et al., 2004). After 1 week, cells were analyzed by means of immunocytochemistry, and 100% of the cells were found to be nestin-positive, whereas >95% strongly expressed Sox2 (Fig. 1D). Also the pluripotency markers Oct4 and Nanog were expressed, although weaker in comparison to Sox2. Oct4 expression was seen mainly in the cytoplasm but not in the nucleus (in accordance with previous findings; Atlasi 2008). Monolayers of nestin-GFP HFNCSCs (p0) were sorted by fluorescence-activated cell sorting (FACS) for GFP after 1 week and analyzed for mRNA expression of pluripotency genes using qRT-PCR (Fig. 1E). Pluripotency gene expression was compared with that in MEFs, ESCs, and previously generated F-iPSCs. Compared to MEFs and ESCs, Sox2 and Nanog are expressed 100-fold higher in HFNCSCs (ESCs, p>0.05; Sox2 and Nanog, p<0.05). HFNCSCs expressed Klf4, Sox2, c-Myc, and Nanog at levels that did not differ significantly from those in ESCs (p>0.05 for all factors); only Oct4 is expressed 100-fold higher in ESCs (p<0.05).
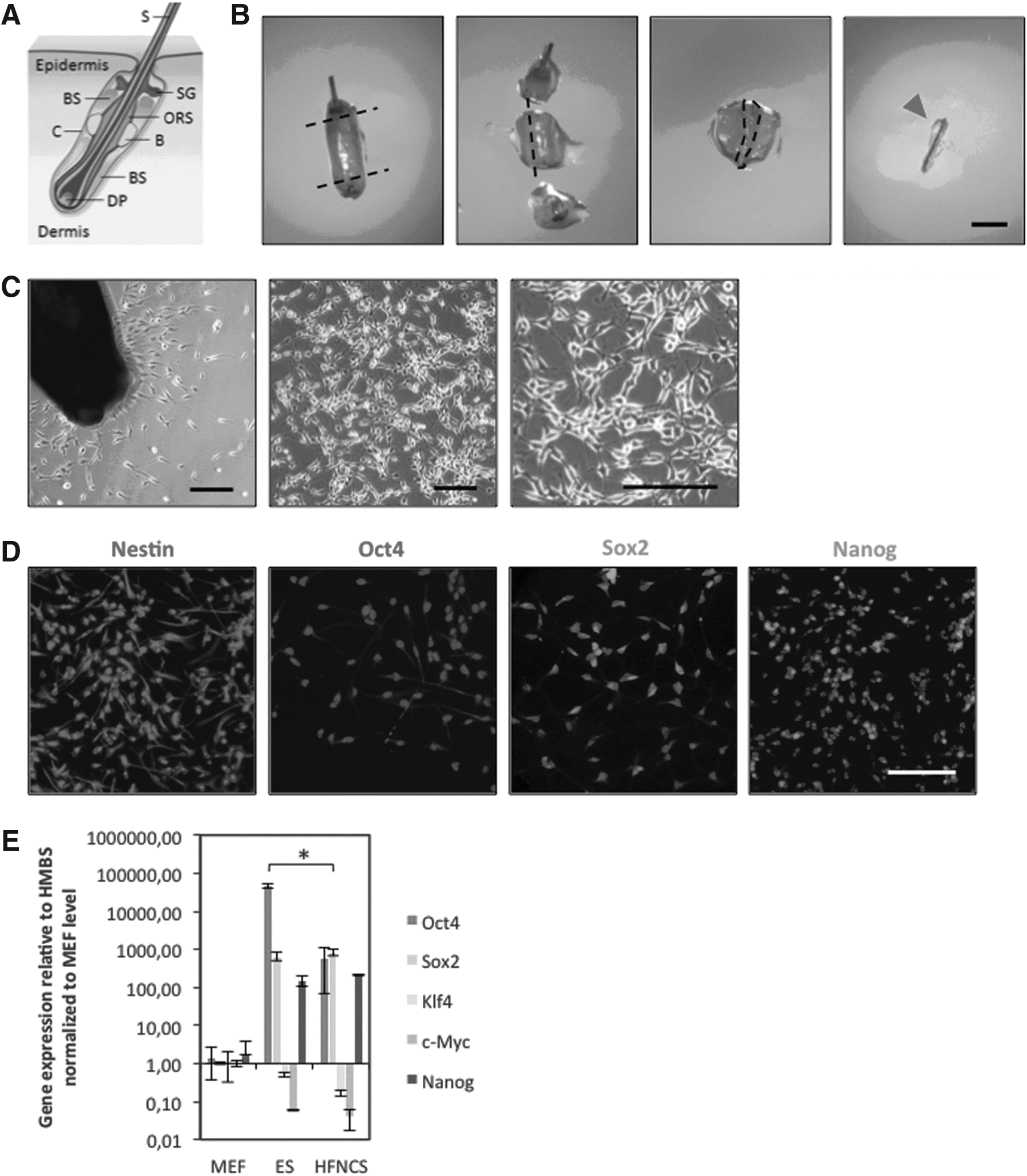
(
Reprogramming of HFNCSCs into iPSCs
In view of the specific endogenous pluripotency expression profiles of the HFNCSCs, we tested whether we could reprogram these cells into iPSCs while omitting the transcription factors already highly expressed by the HFNCSCs (Sox2, Klf4, and/or c-Myc were left out in different combinations as well as altogether; Oct4 was always present). After 4 weeks, no colonies were observed in any of the conditions. Only lentiviral transduction of all four Yamanaka factors in a polycistronic vector resulted in the appearance of iPSC colonies within 5–7 days posttransduction (Fig. 2A). Of 2×104 transduced cells, we counted an average of five colonies per field of view at 100×magnification, or a total of 1125 colonies. Efficiency was calculated at 0.056%, which is slightly higher than efficiencies with fibroblasts and other cell types. Colonies displayed a proper ESC-like morphology with a large nuclear/cytoplasmic ratio and prominent nucleoli. HF-iPSCs were characterized as to their reprogramming and pluripotent characteristics. Like ESCs, HF-iPSCs showed positive AP activity and expression of ESC–associated antigens Oct3/4, Sox2, Nanog, SSEA-1, and SSEA-4 (Fig. 2A, B; results shown for one clone). HF-iPSCs were further characterized by means of quantitative real time-PCR analysis for pluripotency markers and reprogramming factors. High expression levels of Oct4, Sox2, Klf4, and Nanog mRNA were observed. Expression levels of Sox2 and c-Myc were lower (both p<0.05), whereas Oct4, Klf4, and Nanog were higher in HF-iPSCs in comparison to F-iPSCs (Nanog p<0.05, Oct4 and Klf4 p>0.05) (Fig. 2D).
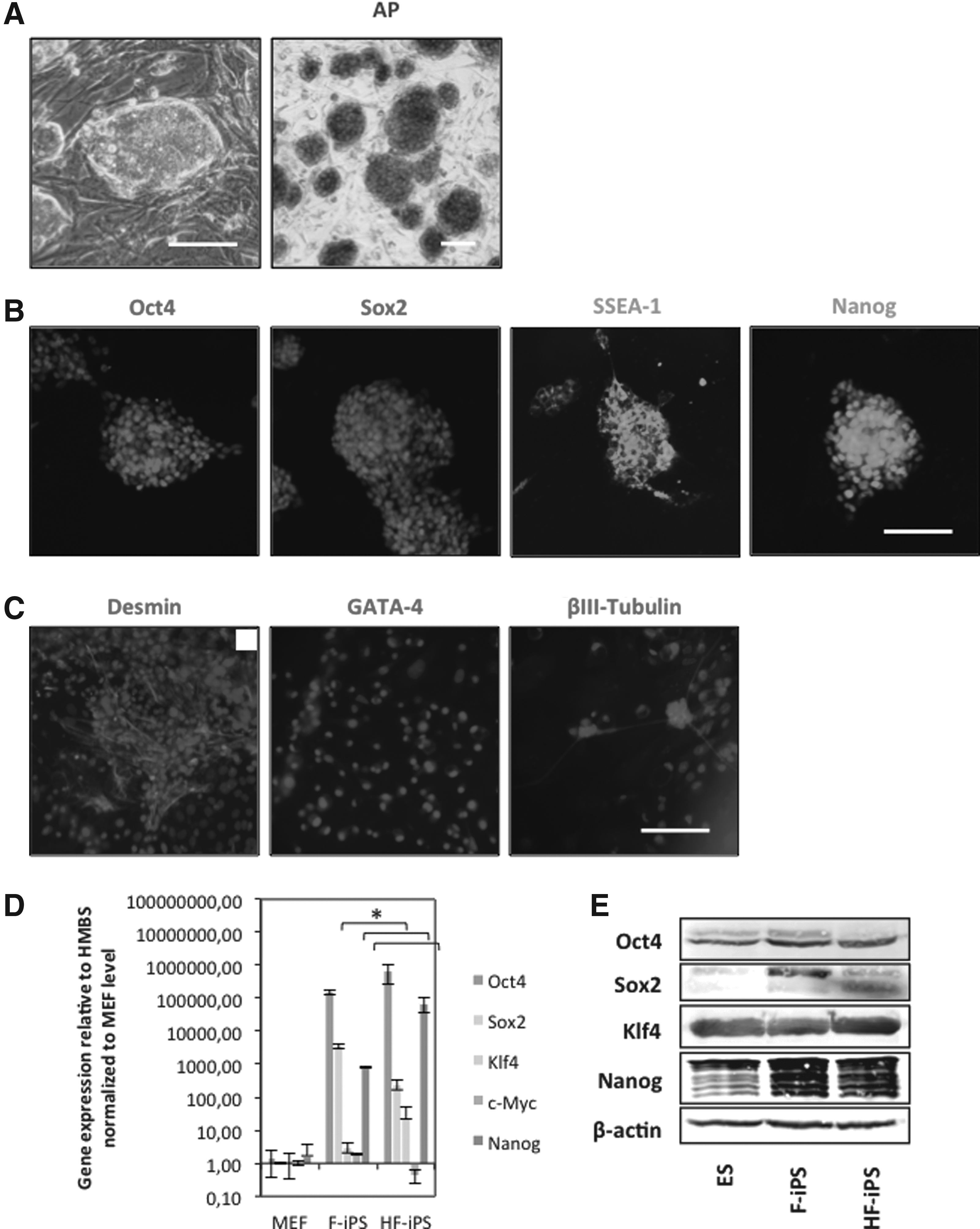
Characterization of HFNCSC-derived mouse iPSCs (HF-iPSCs). (
Expression of pluripotency factors was confirmed on the protein level by means of western blot analysis; expression levels were comparable to those in F-iPSCs, and comparable to or higher (Sox2, Nanog) than levels in ESCs (Fig. 2E). EBs were generated using retinoic acid (RA) according to a −4/+4 protocol, default differentiated in vitro, and stained for markers representative of the three germ layers, i.e., desmin, GATA, and βIII tubulin (Fig. 2C). Positive staining indicated proper default differentiation into the three germ layers.
Discussion
The mammalian hair follicle contains several compartments of stem cells that are responsible for regeneration of these structures during the anagen phase of the hair follicle as well as after injury (for review, see Fuchs and Horsley, 2008; Tiede et al., 2007). A cell population of neural crest origin has been identified in the hair follicle bulge region by the expression of β-galactosidase expression under the Wnt1 promoter in transgenic mouse models (Nagoshi et al., 2008; Sieber‐Blum et al., 2004; Wong et al., 2006). These HFNCSCs express neural crest markers Sox10 and neural crest/neural stem cell marker nestin and can be differentiated toward neural crest lineages such as neurons, smooth muscle cells, and melanocytes in vitro (Fernandes et al., 2004; Hunt et al., 2008; Li et al., 2003; Sieber‐Blum et al., 2004; Toma et al., 2001). Amoh and Hoffman (Amoh et al., 2005; Amoh et al., 2009; Hoffman, 2006) isolated nestin+/keratin 15− multipotent skin-derived cells from the hair follicle bulge of nestin-driven GFP transgenic mice, which could be differentiated into neurons, glia, keratinocytes, smooth muscle cells, and melanocytes in vitro.
Interestingly, some authors have reported the expression of pluripotency-related genes in HFNCSCs. Yu et al. described the presence of nestin-positive cells within the human hair follicle bulge, which in addition were positive for pluripotency factors Nanog and Oct4 (Yu et al., 2006). Sieber-Blum and Hu also showed the expression of (different) pluripotency factors in bulge NCSCs; c-Myc, Klf4, and Sox2 were expressed at similar levels compared to ESCs, whereas Oct4 and Nanog were expressed significantly lower than in ESCs (Sieber-Blum and Hu, 2008). Altogether these studies show that NCSCs persist in the mammalian hair follicle bulge and display several pluripotency-related expression profiles. By means of qRT-PCR, we confirmed that the nestin-expressing NCSCs from the bulge region of the adult mouse whisker follicle (HFNCSCs) co-express high mRNA levels of pluripotency genes Oct4, Sox2, Klf4, and Nanog. Expression of Oct4 was lower than in ESCs, but in contrast to Sieber-Blum and Hu, we showed Nanog expression levels similar to ESCs. Like Yu et al., we were able to detect expression of both Oct4 and Nanog at the protein level.
We hypothesized that because of their endogenous expression of several pluripotency genes, HFNCSCs might be more prone for pluripotency induction, eventually with fewer factors. However, our results show that HFNCSCs can only be reprogrammed using a full set of Yamanaka factors, with an efficiency comparable or only slightly higher than generally found for fibroblasts. Others have shown that endogenous expression levels of pluripotency genes should be able to substitute for ectopic expression, and thus no transgenes would be necessary for iPSC induction in case of high-level expression. The group of Jaenisch and Hochedlinger showed expression levels of Sox2 in neural precursor cells similar to ESC levels, and was able to generate iPSCs retrovirally without exogenous Sox2 (Eminli et al., 2008). Similarly, Kim et al. (2009) showed iPSC induction in SSEA-1–expressing neural stem cells with only Oct4 as transgene. Our data suggest that the levels and activities of the endogenous pluripotency factors were inadequate to contribute significantly to reprogramming of HFNCSCs. This implies that endogenous expression might not be necessarily sufficient for substitution of transgenes, and that ability for reprogramming still depends on the cell type and its epigenetic signature. It appears that cell type is a major factor in determining reprogrammability (Maherali and Hochedlinger, 2008). Unlike, e.g., neural progenitor cells, HFNCSCs or neural crest stem cells in general might be too specified to overcome certain epigenetic barriers restricting reprogramming, and therefore are reprogrammable with only a full set of factors.
Characterization of four-factor derived HF-iPSCs did show ESC-like colony morphology, EB formation and differentiation patterns, and pluripotency marker expression, confirming proper pluripotency induction. The expression of transgene mRNAs was higher than in fibroblast-derived iPSCs, especially for Oct4 and Nanog; this might reflect the high expression levels in the original cells. Assessment of the methylation profiles of the promoter regions of key pluripotency genes would be insightful for determining whether these high expression levels in HF-iPSCs in comparison to F-iPSCs are due to epigenetic differences.
Altogether we have confirmed the pluripotent expression profile of hair follicle bulge NCSCs; however, HFNCSCs could be reprogrammed into fully characterized iPSCs only with a full set of exogenous pluripotency transcription factors (Oct4, Sox2, Klf4, and c-Myc), and without higher efficiency. We conclude that high endogenous levels of Yamanaka factors are no guarantee for facilitated induction of pluripotency.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.