
Abstract
Abstract
β-Cell replacement therapy is a promising field of research that is currently evaluating new sources of cells for clinical use. Pancreatic epithelial cells are potent candidates for β-cell engineering, but their large-scale expansion has not been evidenced yet. Here we describe the efficient expansion and β-cell differentiation of purified human pancreatic duct cells (DCs). When cultured in endothelial growth-promoting media, purified CA19-9+ cells proliferated extensively and achieved up to 22 population doublings over nine passages. While proliferating, human pancreatic duct-derived cells (HDDCs) downregulated most DC markers, but they retained low CK19 and SOX9 gene expression. HDDCs acquired mesenchymal features but differed from fibroblasts or pancreatic stromal cells. Coexpression of duct and mesenchymal markers suggested that HDDCs were derived from DCs via a partial epithelial-to-mesenchymal transition (EMT). This was supported by the blockade of HDDC appearance in CA19-9+ cell cultures after incubation with the EMT inhibitor A83-01. After a differentiation protocol mimicking pancreatic development, HDDC populations contained about 2% of immature insulin-producing cells and showed glucose-unresponsive insulin secretion. Downregulation of the mesenchymal phenotype improved β-cell gene expression profile of differentiated HDDCs without affecting insulin protein expression and secretion. We show that pancreatic ducts represent a new source for engineering large amounts of β-like-cells with potential for treating diabetes.
Introduction
R
Because of its regenerative capacities, the pancreas itself has been extensively studied as a source of progenitors. Various candidates besides the β-cell itself (Russ et al., 2008) have been proposed, including α-cells (Thorel et al., 2010), duct cells (DCs) (Inada et al., 2008), centroacinar cells (Rovira et al., 2010), and acinar cells (Zhou et al., 2008). These studies represent lineage-tracing experiments performed in mice and were convincingly translated in vitro with human tissue only with DCs (Bonner-Weir et al., 2000; Yatoh et al., 2007), reflecting the difficulty of isolating and expanding the other cell types from human pancreas. Human DCs are easily isolated and cultured (Yatoh et al., 2007), but they have not been expanded to the extent needed for cell replacement therapy. This prompted us to focus on how to efficiently derive β-cells from DCs using techniques that would allow unrestricted clinical use and large amounts of cells.
Epithelial cells have limited mitotic activity in vitro, but their expansion can be forced via a phenotype shift. Human β-cells were shown to be able to proliferate in vitro after shifting toward a mesenchymal phenotype through epithelial-to-mesenchymal transition (EMT) (Gershengorn et al., 2004). Lineage-tracing experiments with human cells confirmed the β-cell origin of the mesenchymal cells (Russ et al., 2008) that were able to reacquire β-cell characteristics after the in vitro differentiation protocol (Bar et al., 2012). Similarly, human DCs have β-cell differentiation potential and could theoretically retain their epigenetic memory, even after EMT (Mutskov et al., 2007); thus, they represent an attractive alternative candidate for expansion and differentiation studies.
Here we show a system in which purified human DCs were forced to undergo an EMT that allowed them to proliferate extensively. After expansion, the cells called human pancreatic duct–derived cells (HDDCs) were differentiated in vitro toward β-cell derivatives with a large array of specific marker expression and insulin secretion. This is the first report of viable expansion and β-cell differentiation of human pancreatic exocrine cells using a method compatible with clinical therapy.
Materials and Methods
Cell isolation and culture
Digested pancreatic tissue remaining after islet isolation from 13 human cadaveric donors aged 32–66 years old [body mass index (BMI) 26±2.3 kg/m2] was obtained from Human Islet Isolation teams from San Raffaele Scientific Institute and University of Chicago, after written informed consent and approval by local ethical committees. Within 48 h, human ductal cells were purified using CA19-9 immunomagnetic bead separation, as previously described (Yatoh et al., 2007), with some modifications. In brief, islet-depleted tissue was trypsinized for 20 min, and single cells were incubated with mouse antihuman CA19-9 antibody (1:200, Invitrogen) for 45 min at 4°C. Cells were washed and incubated with 1:5 anti-mouse immunoglobulin G (IgG) microbeads (Miltenyi Biotec) for 30 min at 4°C. Cells were then washed and filtered with 40-μm cell strainers (Falcon) before being positively selected using MACS LS Separation Columns (Miltenyi Biotec) using the manufacturer's instructions. Cell viability was 60–70% as assessed by Trypan Blue dye exclusion. Fresh CA19-9+ DCs were initially seeded on tissue culture treated dishes at 3×105 cells/cm2 in CMRL medium (Gibco) containing 10% fetal bovine serum (FBS; Cellgro) and 1% penicillin/streptomycin (P/S; Cellgro) as a control group or in EGM-2-MV Bulletkit Medium (Lonza) without hydrocortisone to stimulate proliferation. The first medium change was performed after 72–96 h and then twice weekly. Proliferating cells were passaged at 80% confluence using 0.05% trypsin (Cellgro) and subcultured at 5000 cells/cm2.
Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. At each passage, aliquots of HDDCs were suspended in FBS containing 10% dimethyl sulfoxide (Sigma) and frozen in liquid nitrogen. For unselected exocrine tissue culture, tissue was dispersed into four 75-cm2 tissue culture treated flasks and subcultured at 5000 cells/cm2 in 10% FBS-containing 25 mM glucose Dulbecco's Modified Eagle Medium (DMEM; Gibco). Human skin fibroblasts (from healthy volunteers aged 8–35 years old) were cultured in 10% FBS-containing high glucose DMEM. Cumulative population doublings were calculated using the following equation: {[log10(NH)−log10 (NI)]/log10 (2)}, where NI is inoculum number and NH is cell harvest number, as described elsewhere (Bieback et al., 2004). Doubling time was evaluated using the following equation: log10(2)×TH−I/[log10(NH/NI)], where TH − I is time between harvest and inoculum.
Differentiation experiments
β-cell differentiation
Cells were plated at 1.5×104 cells/cm2 on uncoated tissue culture treated six-well dishes. Basal medium (BM) was DMEM/F12 containing 0.1% bovine serum albumin (BSA; Sigma), 10 mM nicotinamide (Sigma), 1×Insulin-Transferrin-Selenium A Supplement (ITS; Gibco), and 1% P/S. To induce β-cell differentiation, HDDCs were submitted to a four-step protocol (R, or recapitulation, protocol) as follows: 100 ng/mL activin A (Peprotech), 1 μM wortmannin (Sigma), and 10 μM SB-216763 (Santa Cruz) (step 1); 5 μM PNU-74654 (Tocris Biosience) (step 2); 5 ng/mL fibroblastic growth factor-2 (FGF-2; Peprotech) and 5 μM forskolin (Calbiochem) (step 3); 2 μM retinoic acid (Sigma) (step 4). Each step was incubated for 3 days. When indicated, cells were incubated 3 days with 1 μM A83-01 (Tocris Bioscience) and 0.5 mM valproic acid (VPA; Sigma) prior to the R protocol (A83-01/VPA–R protocol). Cells were processed for further analyses at the end of the protocol.
Mesodermal differentiation
Cells were plated at 1.5×104 cells/cm2 in uncoated tissue culture treated six-well dishes. For osteogenic differentiation, cells were incubated in DMEM containing 10% FBS, 0.1 μM dexamethasone, 0.05 mM ascorbate, and 10 mM β-glycerophosphate (Sigma) and changed twice a week (Bieback et al., 2004). Extracellular calcium deposition was revealed by Alizarin Red staining after 4 weeks. For adipogenic differentiation (Bieback et al., 2004), cells were incubated with DMEM containing 10% FBS, 1 μM dexamethasone, 0.5 mM isobutylmethylxanthine, 0.2 mM indomethacin, and 10 μg/mL insulin (Sigma) and changed twice a week. After 4 weeks, lipidic vesicles were revealed by Oil Red O staining.
Immunostaining
Cells were fixed in 4% paraformaldehyde (PFA) at room temperature for 20 min and tissues were fixed for 2 h. Freshly isolated DCs were spun, fixed in 4% PFA for 20 min, and pelleted in 2% agar. Tissues and agar pellets were embedded in paraffin. Both cells and sections were washed and then permeabilized with 0.3% Triton X-100 (Sigma) in PBS for 15 min. For sections, antigen retrieval was performed using a pressure cooker in 10 mM citrate acid buffer (pH 6.0). Sections were blocked with 1% donkey serum (Sigma) and cells with 3% BSA (Sigma) for 1 h at room temperature, washed, and incubated at room temperature or at 4°C with primary and secondary antibodies listed in Table S1 (Supplementary Data are available at www.liebertpub.com/cell/). Immunofluorescence used biotin-streptavidin amplification; 4′,6-diamidino-2-phenylindole (DAPI) was used for nuclear staining. Images were acquired in confocal mode on a Zeiss LSM 410 microscope. All immunostaining results were representative of at least three independent experiments. Cell frequencies were evaluated by counting positive cells in four to six different random fields (magnification, 250×) in three different donors. Total cell counts are provided for each experiment. Data are displayed as mean±standard error of the mean (SEM).
Flow cytometry
Cells were trypsinized and suspended at a concentration of 0.5–1×106 cells/50 μL in staining buffer [1% BSA, 0.1% sodium azide, 1×phosphate-buffered saline (PBS)] and then incubated for 30 min at 4°C with monoclonal antibodies directed against a cell-surface antigen (1:50) (Table S1). For intracellular staining, cells were washed and suspended in 250 μL of Fixation/Permeabilization Solution (BD Biosciences) for 20 min at 4°C and then washed in 1×BD Perm/Wash Buffer (BD Biosciences). Cells were immunostained using the biotin-streptavidin method with incubation overnight with 1:100 anti-human insulin, vimentin, or CK19 antibody. Negative controls included unstained cells, control isotypes for membrane staining, and omission of primary antibody for intracellular staining. Cells were analyzed on a BD FACSCanto System (BD Biosciences), and at least 10,000 events were acquired for each sample.
Insulin secretion in vitro
Insulin secretion was measured in culture at the end of differentiation protocols by sequential incubation in 2.8 mM and 20.2 mM glucose in Krebs Ringer Bicarbonate HEPES (16 mM HEPES and 0.1% BSA, pH 7.4) as previously described (Schuppin et al., 1993). Supernatants were stored at −20°C until assayed with a Human Insulin EIA Kit (ALPCO). Three days before the assay, insulin was withdrawn from culture medium, and five washes were performed before applying Krebs solution.
Real time RT-PCR analysis
Total RNA was isolated with an RNeasy Plus Kit (QIAGEN) according to the manufacturer's instructions, and RNA concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). Reverse transcription was carried out using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-Time PCR was performed using specific primers (Table S2), Power SYBR Green PCR Master Mix (Applied Biosystems) and StepOnePlus Real-Time PCR System (Applied Biosystems). Results were analyzed by the comparative ΔCT method (Pfaffl, 2001) and presented in relation to TATA-binding protein (TBP) to avoid misleading fold changes while comparing with low control gene expression levels.
Statistics
Results are expressed as mean±standard error of the mean (SEM) and statistically (*p<0.05, **p<0.01, ***p<0.001) significant differences were assessed using an unpaired Student's t-test.
Results
Freshly isolated human CA19-9+ DCs (fresh DCs) were cultured in EGM-2MV medium containing human epidermal growth factor (hEGF), vascular endothelial growth factor (VEGF), human fibroblast growth factor-basic (hFGFb), long-term analog of insulin-like growth factor-1 (R3-IGF1), ascorbic acid, and FBS. This medium was chosen for its superior induction of proliferation compared to other growth factors, matrices, and culture media. CA19-9+ cells expressed the appropriate receptors for EGM-2MV's endothelial growth-promoting factors (Fig. S1A, B). About 30% of the initially plated cells adhered, and over the first week in culture most cells maintained a cobblestone-like morphology typical of epithelial cells, whereas some were already more elongated without being spindle-shaped (Fig. 1A). Cells proliferated and were passaged at confluence (day 7). With first passage (P1), spindle-shaped cells appeared uniformly throughout culture dishes and proliferated to become the prominent population (Fig. 1B). After P2, cultured cells were uniformly spindle-shaped with no further changes (Fig. 1C). These cells, which we call HDDCs, appeared in all donors (n=13) and after P1 showed a progressive increase of proliferation that peaked at P4–5 and lagged at P9 (Fig. 2A). HDDCs could achieve 17.7±1.8 cumulative population doublings (CPD) in 57.8±5.3 days (n=5) (Fig. 2B). In contrast, no proliferation of spindle-shaped cells was detected when CA19-9+ DCs were cultured in 10% FBS containing CMRL medium or DMEM (n=6). However, when dispersed, unselected exocrine tissue was plated in either of these media, cultures by day 4 contained spindle-shaped cells (n=6) that we called stromal cells (Fig. 1D).
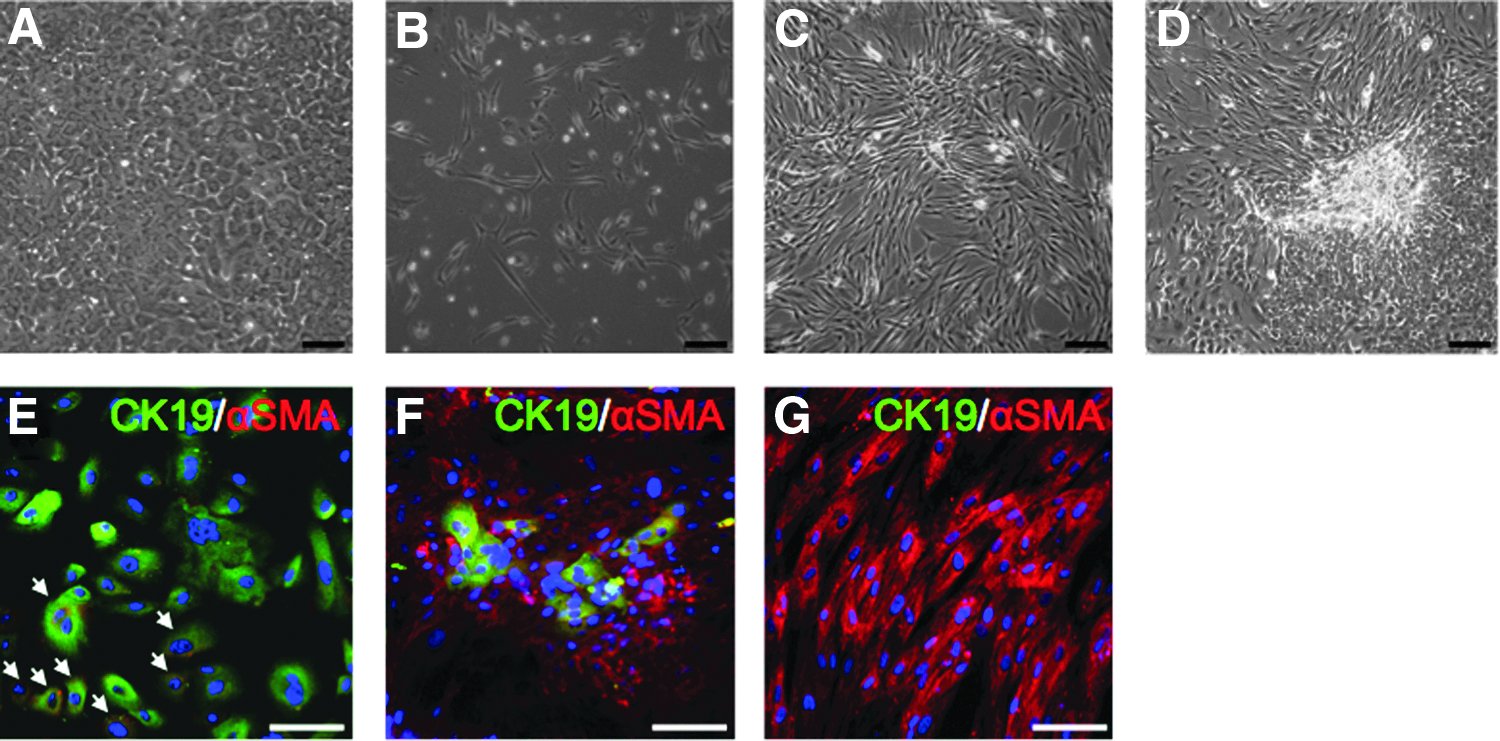
Evolution of HDDC morphology and protein expression profile. (
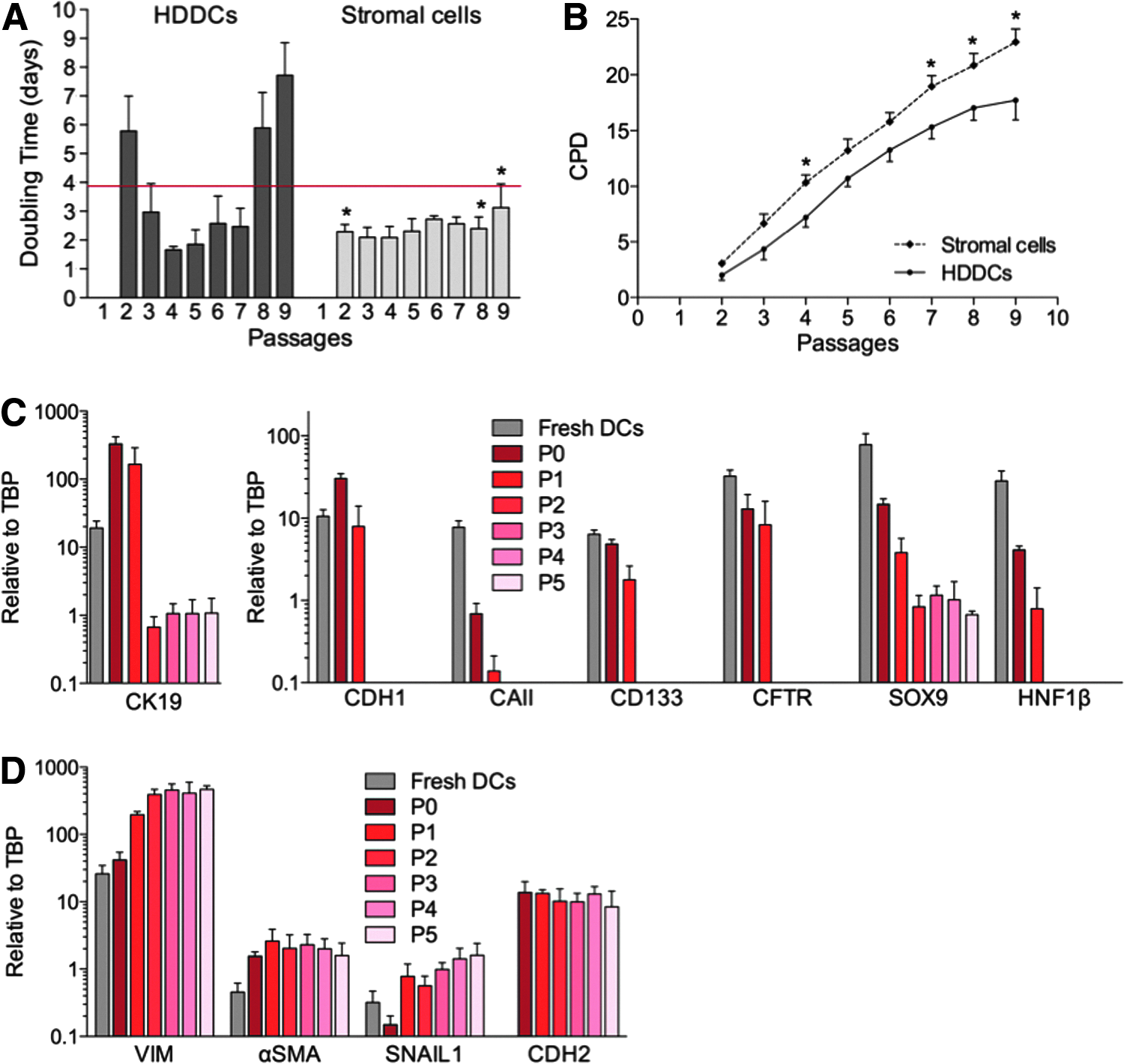
Characterization of HDDCs' proliferation and gene expression profile. (
With passaging, HDDCs lost gene expression of duct marker E-cadherin (CDH1), carbonic anhydrase II (CAII), CD133 (PROM1), CFTR, and HNF1β but maintained low CK19 and SOX9 expression (Fig. 2C). Furthermore, HDDCs upregulated expression of mesenchymal markers vimentin (VIM) and α-smooth muscle actin (αSMA or ACTA2) (Fig. 2D). The EMT marker SNAIL1 was already expressed in fresh DCs (Fig. 2D), suggesting early initiation of EMT (Kalluri and Weinberg, 2009) with dispersion; it was upregulated at P1 compared to P0 (expanded before first passage) (p=0.038), but then varied. The EMT marker N-cadherin (CDH2) was also expressed at P0, but not in fresh DCs (Fig. 2D), possibly reflecting an E- to N-cadherin switch (Zeisberg and Neilson, 2009). Insulin and amylase mRNAs were observed at low levels in fresh DCs, presumably from contaminating cells, but by P1 and P0, respectively, no further contamination of β-cells or acinar cells could be detected (Fig. S1C).
Because HDDCs showed mesenchymal features, we evaluated whether these cells coexpressed CD90, CD73, and CD105, the hallmark marker triad of multipotent mesenchymal stromal cells (MSCs) (Dominici et al., 2006). Using flow cytometry, only a few fresh DCs stained positive for each mesenchymal marker, but by P1 a high proportion of HDDCs expressed the mesenchymal markers CD90, CD105, vimentin, and α-smooth muscle actin (αSMA), whereas only a small proportion expressed CD73 (3.0±1.5% at P3, n=3) (Fig. 3B). Only a small fraction of HDDCs (2.1±1% at P3, n=3) coexpressed the CD90/CD73/CD105 triad, yet this triad was highly expressed in fibroblasts consistent with previous reports (Halfon et al., 2011; Lysy et al., 2007). These results suggest that HDDCs are different from MSCs and fibroblasts. Furthermore, in fresh DCs, no CD90/73/105 coexpression was found, indicating the absence of contaminating MSCs or fibroblasts.
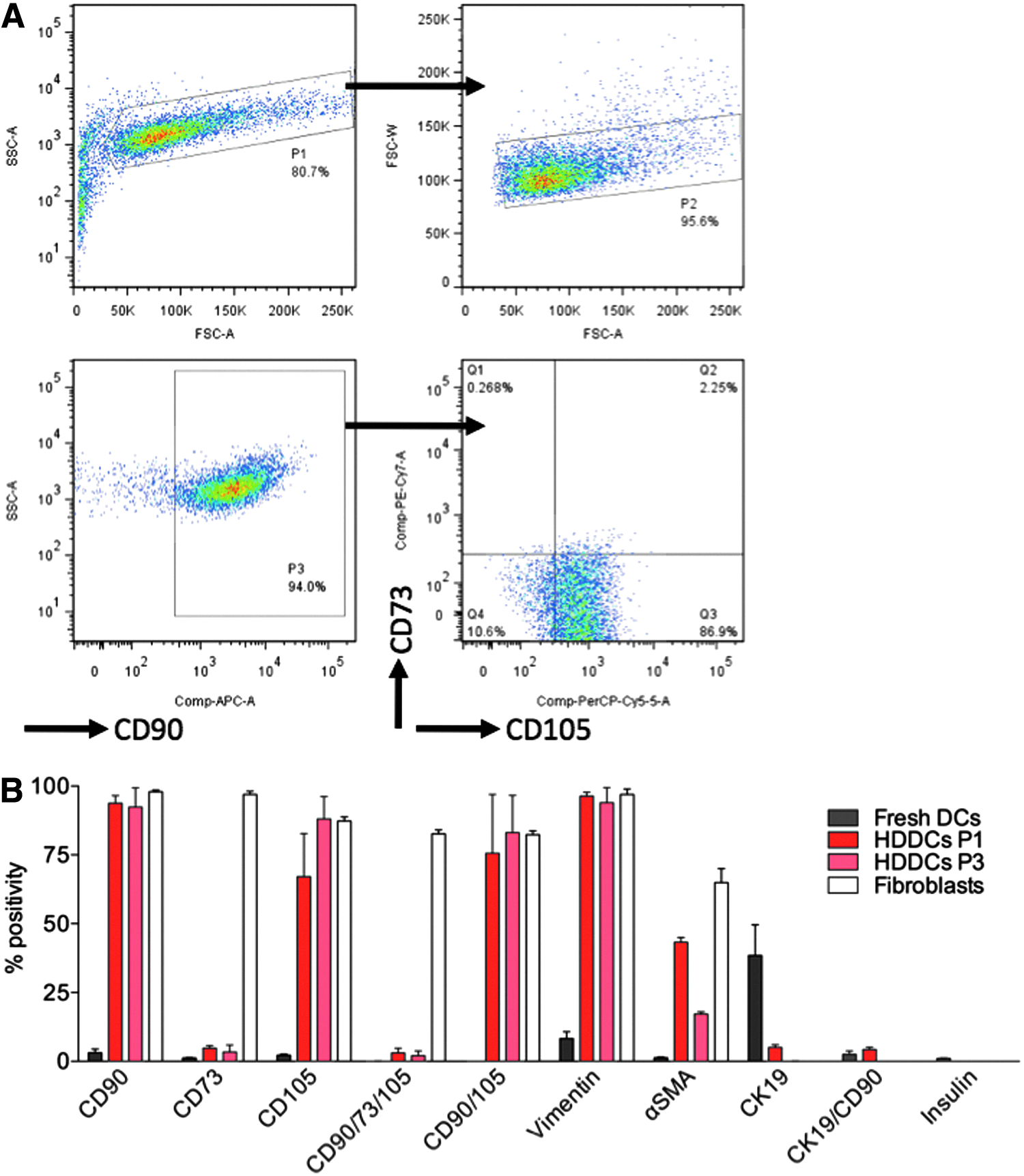
Flow cytometry expression profile of HDDCs. (
Consistent with the mRNA analysis, few insulin+ cells were detected by immunostaining in fresh DC populations with no further trace of insulin after passaging (Fig. 3B). Although CK19 protein expression was downregulated with passaging to undetectable levels at P3, some fresh DC and most P1 CK19+ cells coexpressed CD90, suggesting these cells underwent an EMT immediately after isolation. Immunostaining confirmed that some freshly isolated CK19+ cells expressed vimentin, which is not present in vivo (Bouwens, 1998) (Fig. S1D), and a high proportion of CK19+ cells coexpressed αSMA protein already at P0 (Fig. 1E, arrows). At P1, the proportion of αSMA+ cells was greatly increased, with some remaining CK19+αSMA+ cells (Fig. 1F, all CK19+ cells coexpressed αSMA). From P2 onward, no CK19 protein expression could be detected and virtually all HDDCs expressed αSMA (Fig. 1G) and vimentin (Fig. S1E).
HDDCs, while shifting from the epithelial phenotype, did not acquire the profile of progenitor cells. Pancreatic progenitor–associated transcripts PTF1A, HNF6, HNF4, NKX6.1, NKX2.2, CLDN6, SOX17, GATA4, GATA6, FOXA2, CXCR4, and HEX were all expressed in fresh DCs, but were globally downregulated at P3; only low levels of HNF6, HNF4, and GATA6 mRNAs were expressed at P3 (Fig. S1G). Fibroblasts only expressed GATA4 and GATA6 at low levels; neither cell type expressed OCT4. HDDCs did not shift toward an endothelial phenotype because they expressed no endothelial marker CD31 and low CD105 (also expressed in endothelium) compared to human umbilical vein endothelial cells (HUVECs) (Fig. S1H).
Our data suggest that HDDCs derived directly from DCs through EMT rather than from an outgrowth of contaminating cells. First, HDDCs had a different proliferation profile than stromal cells or fibroblasts. Whereas HDDCs appeared only at P1 and progressively expanded, stromal cells showed robust and consistent growth starting as early as day 4 in initial cultures (P0) and could achieve 22.9±1.2 CPD (n=4) at P9 (Fig. 2A, B), but were able to proliferate up to P12. Stromal cells had a stable doubling time of about 2 days (Fig. 2A) and thus grew faster than fibroblasts. Differences in proliferation between HDDCs and stromal cells widened when HDDCs were cultured in DMEM. Second, the way HDDCs appeared in culture made them unlikely to result from outgrowth of contaminating cells. The CD90/73/105 MSC triad coexpression was undetectable in fresh DCs (Fig. 3), so any contaminating mesenchymal-like cell expressing this triad might be expected to give rise to clonal clusters in culture due to its scarcity (Fig. S1I). However, HDDCs appeared dispersed throughout culture dishes (Fig. S1J) and, when assayed in limiting dilution, lacked the ability to clonally expand (data not shown). This corroborated previous data showing no fibroblastic cells in CA19-9+ cultures (Yatoh et al., 2007). Furthermore, when fresh DCs were incubated with A83-01, an inhibitor of EMT through transforming growth factor-β (TGFβ) signaling blockade, HDDCs did not appear (Fig. S1J).
Additionally, HDDCs differed from stromal cells and fibroblasts (Fig. 4A) in mesenchymal marker gene expression, with significantly lower expression of CD73, CD90, CD105, fibroblast-specific protein 1 (FSP1), and N-cadherin (CDH2) than stromal cells and fibroblasts. Expression of NG2 and PDGFR, recently used with CD146 for isolation of perivascular MSCs from various organs including pancreas (Crisan et al., 2008), was also lower in HDDCs. Only VIM, αSMA, and CD146 were expressed at higher levels in HDDCs than in stromal cells, and only VIM expression was higher in HDDCs than fibroblasts. Importantly, HDDCs also expressed CK19 and SOX9 at low levels, whereas stromal cells or fibroblasts did not. Stromal cells were phenotypically closer to fibroblasts, with their expression profile similar for all analyzed transcripts except αSMA and N-cadherin.
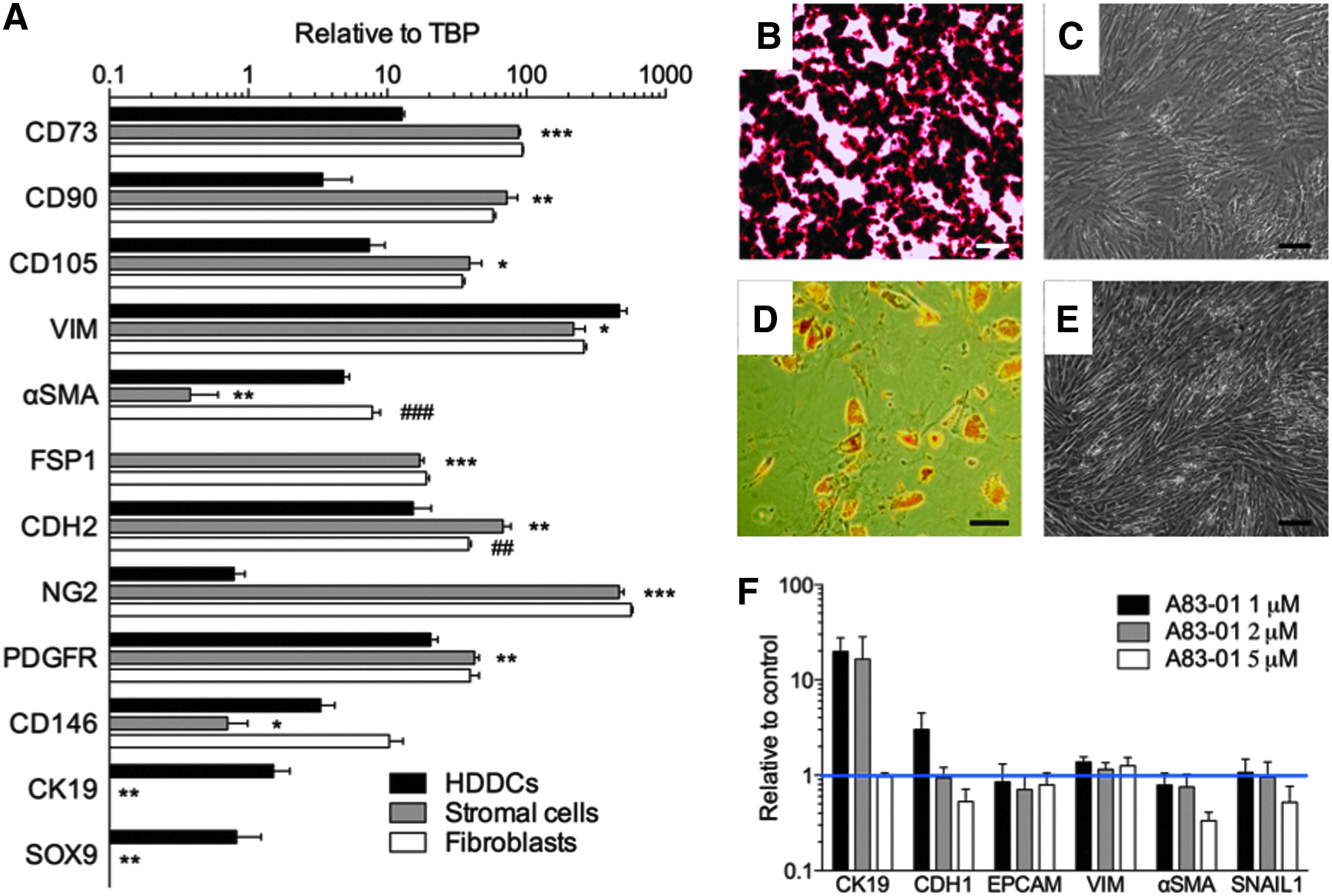
HDDCs differ from pancreatic stromal cells and skin fibroblasts. (
Another difference was the capacity to differentiate into bone, fat, and cartilage, which is the stem cell hallmark of MSCs (Salem and Thiemermann, 2010). HDDCs lacked the potential to fully differentiate into osteocyte or adipocyte; they did not secrete calcium matrix (Fig. 4C) nor accumulate cytoplasmic lipid droplets (Fig. 4E). Stromal cells similarly lacked mesodermal differentiation (not shown). Thus, because they retained epithelial markers and had blunted mesenchymal features (Table 1), HDDCs are distinct from stromal cells, fibroblasts, and MSCs. To evaluate whether HDDCs could reverse their phenotype and regain epithelial features, we incubated HDDCs with the EMT inhibitor A83-01. The cells upregulated CK19 and E-cadherin (CDH1) mRNA expression (Fig. 4F), without modifying their morphology, suggesting they may retain epigenetic characteristics of epithelial cells.
DC, duct cells; MSCs, mesenchymal stromal cells; HDDCs, human pancreatic duct-derived cells; αSMA, alpha-smooth muscle actin; P0, passage zero.
To evaluate the β-cell differentiation potential of HDDCs, we designed a differentiation protocol (called R for recapitulation) recapitulating pancreatic development (Champeris Tsaniras and Jones, 2010) after screening for chemicals and growth factors. Differentiation experiments were performed on HDDCs from P2 to P5 devoid of β-cell contamination. When analyzed after incubation with the differentiation protocol, HDDCs significantly enhanced the expression of insulin, PDX1, MAFA, NEUROD, NKX6.1, GLUT2 (SLC2A2), and glucokinase (GCK) mRNAs whereas glucagon (GCG) expression was very low (Fig. 5A). Control experiments using fresh human DCs, stromal cells, fibroblasts, PANC-1 cells, or umbilical cord matrix stem cells incubated with this protocol failed to upregulate insulin mRNA expression (data not shown).
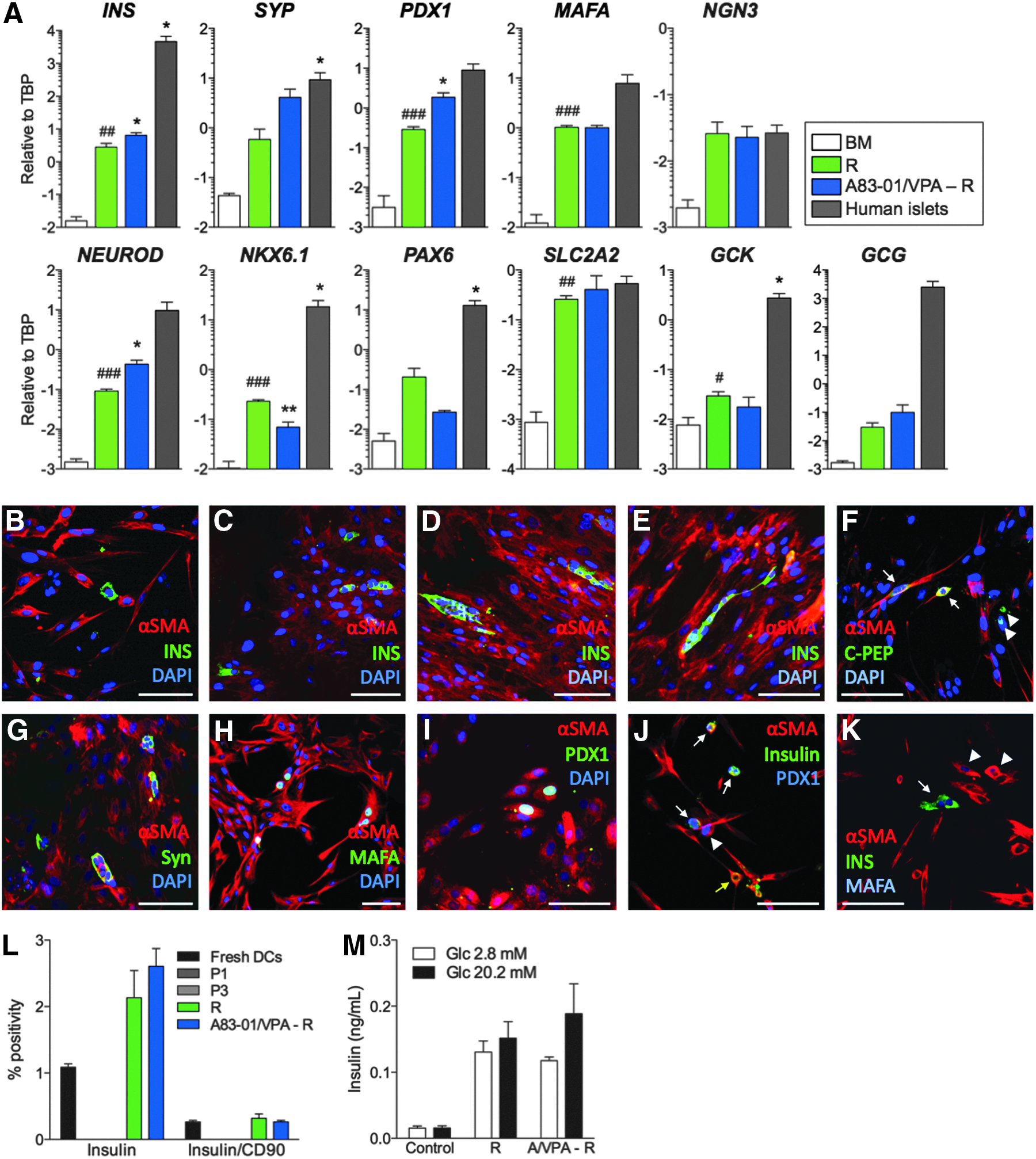
In vitro β-cell differentiation of HDDCs. (
To further improve the differentiation protocol, we tested the hypothesis that a gain of epithelial phenotype in HDDCs could enhance their β-cell differentiation. This hypothesis was based on our data that HDDCs could upregulate their epithelial phenotype after EMT inhibition and that MET was required to reprogram fibroblasts into iPSCs (Li et al., 2010b). For this protocol, we incubated HDDCs with A83-01 and VPA, a histone deacetylase inhibitor shown to synergize with RepSox EMT inhibitor (Ichida et al., 2009), for 3 days prior to our R protocol. Compared to the R protocol, these HDDCs had higher INS, PDX1, and NEUROD mRNA expression without significantly different expression of other analyzed markers, including glucagon (Fig. 5A). These data suggest that reducing the mesenchymal phenotype of HDDCs furthers differentiation.
By immunostaining, R-differentiated HDDC populations contained 1.8±0.4% insulin+ cells (n=3) (Fig. 5B–E), as compared to HDDCs incubated during 14 days in basal medium that did not contain insulin+ cells. Even though A83-01/VPA-R–induced HDDCs had higher insulin mRNA levels than after the R protocol, the insulin+ cell proportions after this protocol (2.0±0.3%, n=3) did not differ from R (data not shown). In all differentiation experiments, insulin+ cells appeared as single cells, as small clusters, or remained spindle-shaped and coexpressed αSMA, showing a persistent mesenchymal phenotype. HDDCs incubated with the R protocol also acquired protein expression of C-peptide (Fig. 5F), synaptophysin (Fig. 5G), MAFA (Fig. 5H, K), and PDX1 (Fig. 5I, J). None of the insulin+ cells coexpressed glucagon, pancreatic polypeptide, or somatostatin (data not shown). Flow cytometry confirmed these proportions of insulin+ cells for R and A83-01/VPA–R protocols with none in P1 or P3 HDDCs (Fig. 5L). This analysis also revealed that even though CD90 was highly expressed in naive HDDCs (Fig. 3), only a small proportion of insulin+ cells still coexpressed CD90. Importantly, differentiated HDDC from these two protocols secreted insulin under basal conditions (2.8 mM glucose) but were not responsive to increased (20.2 mM) glucose stimulation (Fig. 5M).
Discussion
We describe for the first time the isolation, characterization, and differentiation of expanded HDDCs. HDDCs were reliably isolated (100% of donors) and highly expandable such that if 100×106 CA19-9+ cells were isolated from a donor (a usual yield), 1011 HDDCs could be harvested at P5, after 1 month. HDDCs can be cryopreserved without phenotypic alteration after thawing (data not shown) and be used for differentiation and transplantation. These features make HDDCs available for clinical procedures and represent a major improvement compared to previously described pancreatic progenitor cells lacking significant expansion capacities (Seaberg et al., 2004; Hao et al., 2006; Ramiya et al., 2000). With these cells, we generated insulin-expressing β-cells that acquired global β-like cell expression pattern.
During expansion, HDDCs changed from a cobblestone to spindle-shaped morphology and underwent an EMT with coexpression of mesenchymal and epithelial markers. EMT has been shown to be a mechanism allowing proliferation of mesenchymal derivatives from ex vivo cultured human islets (Gershengorn et al., 2004; Russ et al., 2008; Russ et al., 2009), a feature not observed in mouse (Atouf et al., 2007; Weinberg et al., 2007). In those human studies, EMT cells derived directly from the β-cells. Similarly, our evidence strongly supports that HDDCs arose directly from CA19-9+ ductal cells rather than from contamination (Table 1). The HDDCs phenotype corresponded to a partial EMT, different from classical in vivo EMT models (Zeisberg and Neilson, 2009) because it appeared de novo in specific culture conditions. Partial EMT has also been described during embryogenesis (Leroy and Mostov, 2007). The stimulation of epithelial marker expression in HDDCs after either incubation with A83-01 or downregulation of vimentin or SNAIL1 expression provided indirect evidence of the incompleteness of EMT. EMT-produced HDDCs were not full-fledged mesenchymal cells because their expression profile differed from that of stromal cells, fibroblasts, or MSCs for the most representative mesenchymal markers (Garcia-Gomez et al., 2006) and because HDDCs also lacked the MSCs' characteristic of mesodermal differentiation. This is consistent with the reported absence of mesodermal differentiation of mesenchymal lineages arising from human islets (Russ et al., 2009).
We defined culture conditions that induced β-cell differentiation in HDDCs. The R protocol was designed to recapitulate pancreatic development (Champeris Tsaniras and Jones, 2010) because β-cell neogenesis from DCs occurs in rodents after pancreatic injury by recapitulating development (Bonner-Weir et al., 1993; Li et al., 2010b). The first step included an activator of Wnt signaling (SB-216763) and activin A, which promotes definitive endoderm differentiation when coincident with wortmannin-induced low phosphoinositide 3-kinase (PI3K) signaling. HDDCs were then exposed to Wnt pathway inhibitor PNU-74654; the third step inhibited the sonic hedgehog pathway using bFGF and protein kinase A activators forskolin or isobutylmethylxanthine (IBMX). Cells were finally incubated with bFGF and forskolin, which synergized. The protocol ended with retinoic acid, a Notch inhibitor that proved superior to DAPT.
HDDCs showed the potential to reverse to their epithelial phenotype, so we hypothesized that downregulation of mesenchymal markers would enhance their plasticity. Inhibition of TGF-β signaling enhanced expression of β-cell markers. Although neither insulin protein expression nor secretion improved compared to the R protocol, up to 3.1% insulin+ cells could be detected. By comparison, D'Amour et al. obtained with human ESCs up to 12% insulin+ cells (average 7%) (D'Amour et al., 2006). Even though our system yielded a lower proportion of insulin+ cells, it remains nevertheless competitive because HDDCs can be produced in large amounts without ethical or evident safety issues. HDDCs arise from EMT, a process involved in tumor formation, therefore full characterization of the genomic stability and in vivo behavior of the cells will be mandatory. Reports of human islets-derived EMT cells that are more comparable to our cells were discrepant in obtaining β-cells after differentiation, ranging from absence of differentiation (Kayali et al., 2007) to noticeable amounts (up to 25%) of insulin+ cells (Bar et al., 2012; Ouziel-Yahalom et al., 2006). Variability in differentiation protocols [cell aggregation only (Kayali et al., 2007), growth factors (Bar et al., 2012; Ouziel-Yahalom et al., 2006)] and contaminating β-cells in subcultures (P5) (Ouziel-Yahalom et al., 2006) could at least partially account for the observed differences. However, further research is awaited to confirm the encouraging new data with islet-derived EMT cells (Bar et al., 2012). Reprogramming variability is also observed in exocrine cell populations. In fact, the incapacity for β-cell differentiation of contaminating stromal cells arising from unselected exocrine cultures was recently described using transcription factor-based protocols (Lima et al., 2013). Hence, we were able to derive insulin+ cells from HDDCs but not from freshly plated DCs or from stromal cells.
In our system, the question arises whether the β-cells found after differentiation were residual contaminating cells carried through the culture. Whereas initial freshly isolated CA19-9+ cell populations had occasional acinar and β-cells but no mesenchymal cells, our characterization was performed after extensive proliferation and no insulin mRNA was detected from P1. Simple mathematical calculations ruled out the possibility. Approximately 2% insulin-producing cells are observed in HDDCs differentiated at P5 with either R or A83-01/VPA-R protocols. HDDCs from P0 to P5 achieve 10.7 population doublings, so if we started with 1×106 CA19-9+ cells at P0, we could obtain 2.5×109 HDDCs at P5 and harvest 53×106 insulin+ cells. For residual islet or DCs to be the origin of those cells, they would need to perform at least 5.7 population doublings in 32 days, while maintaining their epithelial phenotype and 100% efficiency of β-cell differentiation, all of which is quite unlikely.
Our study shows that HDDCs are potential candidates for β-cell replacement therapy for diabetes. These cells are easily harvested and can be reliably expanded such that one human donor could potentially serve for several transplant recipients. After in vitro differentiation, HDDCs generate β-like cells with insulin secretion capacities. Additional in vivo studies are required to evaluate the potential of HDDCs as a therapy for diabetes.
Footnotes
Acknowledgments
The authors thank J. Ravau (Pôle Pédiatrie) for his expert technical assistance.
This study was supported by grants from Belgian Study Group for Pediatric Endocrinology (BSGPE), Institut de Recherche Clinique et Expérimentale (IREC), Fonds National de la Recherche Scientifique (FNRS), and Université Catholique de Louvain (UCL). Dr. Bonner-Weir was supported by National Institutes of Health (NIH) grants DK044523, DK074879, and DK036836.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.