
Abstract
Abstract
Synthetic modified mRNA molecules encoding pluripotency transcription factors have been used successfully in reprogramming human fibroblasts to induced pluripotent stem cells (iPSCs). We have applied this method on bone marrow–derived mesenchymal stromal cells (BM-MSCs) obtained from a patient with β-thalassemia (β-thal) with the aim to generate trangene-free β-thal-iPSCs. Transfection of 104 BM-MSCs by lipofection with mRNA encoding the reprogramming factors Oct4, Klf4, Sox2, cMyc, and Lin28 resulted in formation of five iPSC colonies, from which three were picked up and expanded in β-thal-iPSC lines. After 10 serial passages in vitro, β-thal-iPSCs maintain genetic stability as shown by array comparative genomic hybridization (aCGH) and are capable of forming embryoid bodies in vitro and teratomas in vivo. Their gene expression profile compared to human embryonic stem cells (ESCs) and BM-MSCs seems to be similar to that of ESCs, whereas it differs from the profile of the parental BM-MSCs. Differentiation cultures toward a hematopoietic lineage showed the generation of CD34+ progenitors up to 10%, but with a decreased hematopoietic colony-forming capability. In conclusion, we report herein the generation of transgene-free β-thal-iPSCs that could be widely used for disease modeling and gene therapy applications. Moreover, it was demonstrated that the mRNA-based reprogramming method, used mainly in fibroblasts, is also suitable for reprogramming of human BM-MSCs.
Introduction
T
However, all methods that use DNA constructs carry the risks associated with the integration of exogenous genetic material into the genome of the target cells. Thus, the most promising reprogramming methods for generating iPSCs suitable for clinical application are those that use either protein or mRNA or micro RNA (miRNA) molecules. Although induction of pluripotency in human cells by direct delivery of four reprogramming proteins (Oct4, Sox2, Klf4, and c-Myc) or by transfection of mature miRNAs has been reported, the generation of iPSCs by both strategies was very slow and inefficient (Kim et al., 2009; Miyoshi et al., 2011). In contrast, the delivery by lipofection of synthetic mRNA molecules encoding reprogramming transcription factors has been shown to be highly effective in reprogramming of human fibroblasts in terms of high reprogramming efficiency and rapid kinetics with which iPSCs were generated. More specifically, the mRNA-based reprogramming protocol developed by Warren et al. (2010) is two times faster and 35-fold more efficient in reprogramming human fibroblasts than the viral one. Nevertheless, how efficient this method is on other cell types such as bone marrow–derived mesenchymal stromal cells (BM-MSCs) remains to be shown. In the present study, we aimed at generating transgene-free β-thal-iPSC lines through reprogramming of BM-MSCs by synthetic modified mRNA molecules.
Materials and Methods
Cell culture
BM was harvested from the posterior iliac crest for an autologous backup BM graft of a patient with β-thal major (IVSI-n6T→C/−87C→T, HBB:c.[92+6T→C]+[-137C→T]), who would undergo hematopoietic stem cell transplantation from her human leukocyte antigen (HLA)-matched sibling. A sample of 15 mL was used after written informed consent from the parents. The research protocol was approved from the Ethical Committee of the Aghia Sophia Children's Hospital (Athens, Greece). Mononuclear cells were isolated from BM by density gradient centrifugation on Ficoll and suspended in Dulbecco's Modified Eagle Medium (DMEM; GlutaMAX, Invitrogen, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (FBS; Stemcell Technologies, Vancouver, BC, Canada) and 20 ng/mL basic fibroblast growth factor (bFGF; R&D Systems, Minneapolis, MN, USA), and placed into a 75-cm2 flask. The adherent cells were proliferated and passaged two times to obtain a population enriched in MSCs. Cultured cells were analyzed by flow cytometry (Epics XL-MCL, Beckman Coulter Inc.) and were stained with the following human specific monoclonal antibodies: CD29-FITC, CD90-PE, CD44-FITC, CD105-FITC, CD45-PC5, and CD31-PE.
BJ human foreskin fibroblasts were purchased from Stemgent (Cambridge, MA, USA) and human neonatal foreskin fibroblasts (NuFF) were obtained from GlobalStem (Gaithersburg, MD, USA). Skin fibroblasts were maintained in DMEM GlutaMAX containing 10% FBS.
Generation of iPSCs with synthetic mRNA
Synthetic mRNAs (Stemgent, Cambridge, MA, USA) of five transcription factors, Oct4, Klf4, Sox2, c-Myc, and Lin28 (in a stoichiometry of 3:1:1:1:1, respectively), were used for the reprogramming of BM-MSCs to pluripotency. These mRNAs incorporate modifications designed to overcome innate antiviral responses. The mRNA reprogramming was performed according to the manufacturers' recommendations. Briefly, NuFF cells were irradiated (40 Gy) and seeded at 2.5×104 /cm2. The following day, BM-MSCs were plated to a density of 2500/cm2 in duplicate cultures. After 2 days, the culture medium was replaced with Pluriton medium (Stemgent) that was supplemented with 200 ng/mL B18R interferon inhibitor (eBioscience, San Diego, CA, USA), and mRNA transfections were initiated with RNAiMAX Transfection Reagent (Invitrogen) cationic lipid delivery vehicles and repeated every 24 h for 18 days. Pluriton medium was replaced daily, 4 h after transfection. The daily RNA dose applied was 1200 ng per well (six-well plate format). mRNA encoding nucleus-localizing green fluorescent protein (nGFP) was also included in the mRNA cocktail as a marker for monitoring transfection efficiency. Moreover, a GFP mRNA dose of 500 ng was delivered to a well of a 12-well plate using RNAiMAX Trasnfection Reagent to test the transfection efficiency for BM-MSCs. For the positive control, we performed a reprogramming experiment with BJ human fibroblasts at the same time.
Culture and expansion of iPSCs
Each iPSC colony was picked manually and transferred to an individual well of a 12-well plate precoated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) in mTeSR medium (Stemcell Technologies). For the first few passages, the cells were passaged mechanically and further expanded by enzymatic passaging with dispase (Stemcell Technologies).
Human ESC line
The HUES-9 cell line was used in this work and served as a positive control in comparative molecular and culture studies assessing pluripotency and differentiation potential of iPSCs, respectively. HUES-9 cells were grown on plates precoated with Matrigel in mTeSR medium.
Flow cytometry analysis for expression of pluripotency markers
Oct3/4 and stage-specific embryonic antigen-4 (SSEA-4) primary antibodies (Stemcell Technologies) were used to characterize iPSCs by flow cytometry. Staining with unconjugated mouse isotypic controls was performed in parallel. The secondary antibody used was fluorescein isothiocyantate (FITC)-conjugated goat anti-mouse immunoglobulin M (IgM). Briefly, for surface SSEA-4 staining, cell were first incubated in a final 1:10 dilution of primary antibody for 30 min, subsequently washed and blocked with 10% FBS/phosphate-buffered saline (PBS), and, finally, incubated with secondary antibody for 30 min. For intracellular Oct3/4 staining, IntraPrep Permeabilization Reagents (Beckman Coulter Inc., Nyon, Switzerland) were additionally used. Namely, iPSCs were first fixed in paraformaldehyde solution for 15 min at room temperature. After washing with 10% FCS/PBS, cells were initially permeabilized in saponin solution and labeled with the primary antibody for Oct3/4, at a final dilution of 1:10. After incubation for 30 min, cells were washed in 10% FBS/PBS and incubated with saponin solution and a final dilution of 1:10 of the secondary antibody. Samples were suspended in PBS and analyzed by flow cytometry (FC500, Beckman Coulter Inc.).
RT-PCR analysis
Total mRNA was extracted from iPSC pellets using an RNeasy Plus Mini-kit (Qiagen GmbH, Hilden, Germany) and reverse transcribed into cDNA using a SuperScript Reverse Transcriptase Kit (Invitrogen). The resulting cDNA was then used as a template for detecting pluripotency marker gene expression of OCT3/4, REX-1, SOX2, and NANOG. Results were compared to expression levels of housekeeping gene GAPDH (glyceraldehyde 3-phosphate dehydrogenase).
DNA array comparative genomic hybridization analysis
Total genomic DNA was extracted with Qiagen Blood Mini-kit. 800ng of the extracted gDNA was amplified and labeled using the SureTag DNA Labeling Kit, Two Colors (Agilent Technologies, Santa Clara, CA, USA). The labeled DNA was hybridized to Agilent SurePrint G3 Human 4x180k CGH+SNP microarrays. The 4x180K CGH+SNP platform is composed of 110,712 [comparative genomic hybridization (CGH)]+59,647 [single-nucleotide polymorphism (SNP)] 60-mer oligonucleotide probes with 25.3-kb overall median probe spacing (5-kb in ISCA regions). Agilent Feature Extraction Image Analysis Software (v. 10.7.3) was used to extract data from raw microarray image files. Data visualization and analysis was performed using Agilent CytoGenomics (v. 2.7) software. For the location of genes [annotated against National Center for Biotechnology Information (NCBI) build 37, hg19] in the deleted/duplicated genomic segments the University of California Santa Cruz (UCSC; http://genome.ucsc.edu/) and the Database of Genomic Variants (http://projects.tcag.ca/variation/) were used.
Gene expression analysis
Total RNA was extracted with a Qiagen RNeasy Mini Kit. A 25-ng amount of total RNA was amplified and labeled using the Agilent Low Input Quick Amp Labeling Kit, One-Color. The labeled RNA was hybridized to Agilent SurePrint G3 Human Gene Expression 8x60K Microarrays. The specific platform is composed of 50,599 60-mer oligonucleotide probes. Agilent Feature Extraction Image Analysis Software (v. 10.7.3) was used to extract data from raw microarray image files. Data visualization and analysis was performed using GeneSpring GX (v. 11.0) software.
In vitro differentiation
For embryoid body (EB) formation, iPSCs were treated with 1 mg/mL dispase and transferred to six-well low-attachment plates in differentiation medium composed of knockout DMEM supplemented with 20% knockout serum replacement, 1×nonessential amino acids, 1 mM
In vivo teratoma formation
For the teratoma formation assay, iPSCs from one T25 flask were harvested by dispase treatment (1 mg/mL) and resuspended in DMEM/F12 and Matrigel at a ratio 2:1. The cell mixtures were subcutaneously injected into 6-week-old severe combined immunodeficient (SCID) mice. After 7–11 weeks, the resultant tumors were dissected, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. After staining with Hematoxylin & Eosin, the sections were examined for the presence of tissues derived from the three germ layers.
Hematopoietic differentiation
The hematopoietic differentiation of BM-MSCs was investigated in duplicate adherent cell cultures using the recombinant protein-based, animal product-free medium STEMdiff APEL (Stemcell Technologies). Briefly, 3 days after passaging iPSCs (day 0), mTeSR medium was replaced with STEMdiff APEL medium, supplemented with 30 ng/mL vascular endothelial growth factor (VEGF), 30 ng/mL bone morphogenetic protein-4 (BMP-4), 40 ng/mL stem cell factor (SCF), and 50 ng/mL Activin A (all from R&D Systems). On day 4, this medium was removed and replaced with STEMdiff APEL medium supplemented with 300 ng/mL SCF, 300 ng/mL Flt-3 ligand, 10 ng/mL interleukin-3 (IL-3), 10 ng/mL IL-6, 50 ng/mL granulocyte colony-stimulating factor (G-CSF), and 25 ng/mL BMP-4 (all from R&D Systems). The medium was changed every 3 days. On day 13, cells were treated with TrypLE Select (Invitrogen) and analyzed for expression of hematopoietic markers CD34 and CD45 by flow cytometry. Single cells were also cytospun on slides and stained with May Gruenwald–Giemsa. For the colony forming unit (CFU) assay, 1–2×105 cells were plated in duplicate 35-mm tissue culture dishes containing 1 mL of MethoCult H4230 (Stemcell Technologies) supplemented with 100 ng/mL SCF, 5 U/mL erythropoietin (EPO), 50 ng/mL IL-3, 50 ng/mL G-CSF, 50 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; R&D Systems) and cultured for 13 more days.
Results
Generation of β-thal-iPSCs from BM-MSCs
We first established a culture of BM-MSCs obtained from the thalassemic patient as described in Materials and Methods. After two passages in vitro, the cells displayed a fibroblast-like morphology. Flow cytometric analysis of cultured cells demonstrated expression of cell-surface markers typical of cultured MSCs, such as CD44, CD105, CD29, and CD90, and did not express the markers CD45 and CD31 specific for hematopoietic and endothelial cells, respectively.
To reprogram these cells, MSC cultures containing 104 cells were transfected daily with a five factor–modified mRNA cocktail for 18 days. Loosely packed colonies began to emerge as early as day 11 of transfection, whereas typical iPSC colonies could be seen at day 15. These colonies reached the optimum size to be picked up around days 18–21. An outline of the reprogramming approach is shown in Figure 1A. To test the transfection efficiency of mRNAs for BM-MSCs, we analyzed the GFP expression by flow cytometry. Twenty-four hours posttransfection, we detected 85% GFP-positive cells (Fig. 1B). To compare the efficiency of iPSC derivation from BM-MSCs directly, we performed an experiment in which the reprogramming efficiency of this method was also defined in BJ fibroblasts. The average number of PSC-like colonies developed from two wells was five for BM-MSCs and about 150 for BJ fibroblasts, corresponding to a reprogramming efficiency of 0.05% and 3.7%, respectively. These results demonstrate a 74-fold lower iPSC conversion efficiency in BM-MSCs than in BJ fibroblasts. After 2 days posttransfection, when the BM-MSC–derived iPSC colonies had a good-looking PSC-like morphology and were sufficiently expanded in size, we manually picked three colonies and replated each colony to an individual well precoated with Matrigel in mTeSR medium for maintenance in a feeder-free, xeno-free system. All three clones were expanded in iPSC lines and retained their undifferentiated state for more than 3 months in culture.
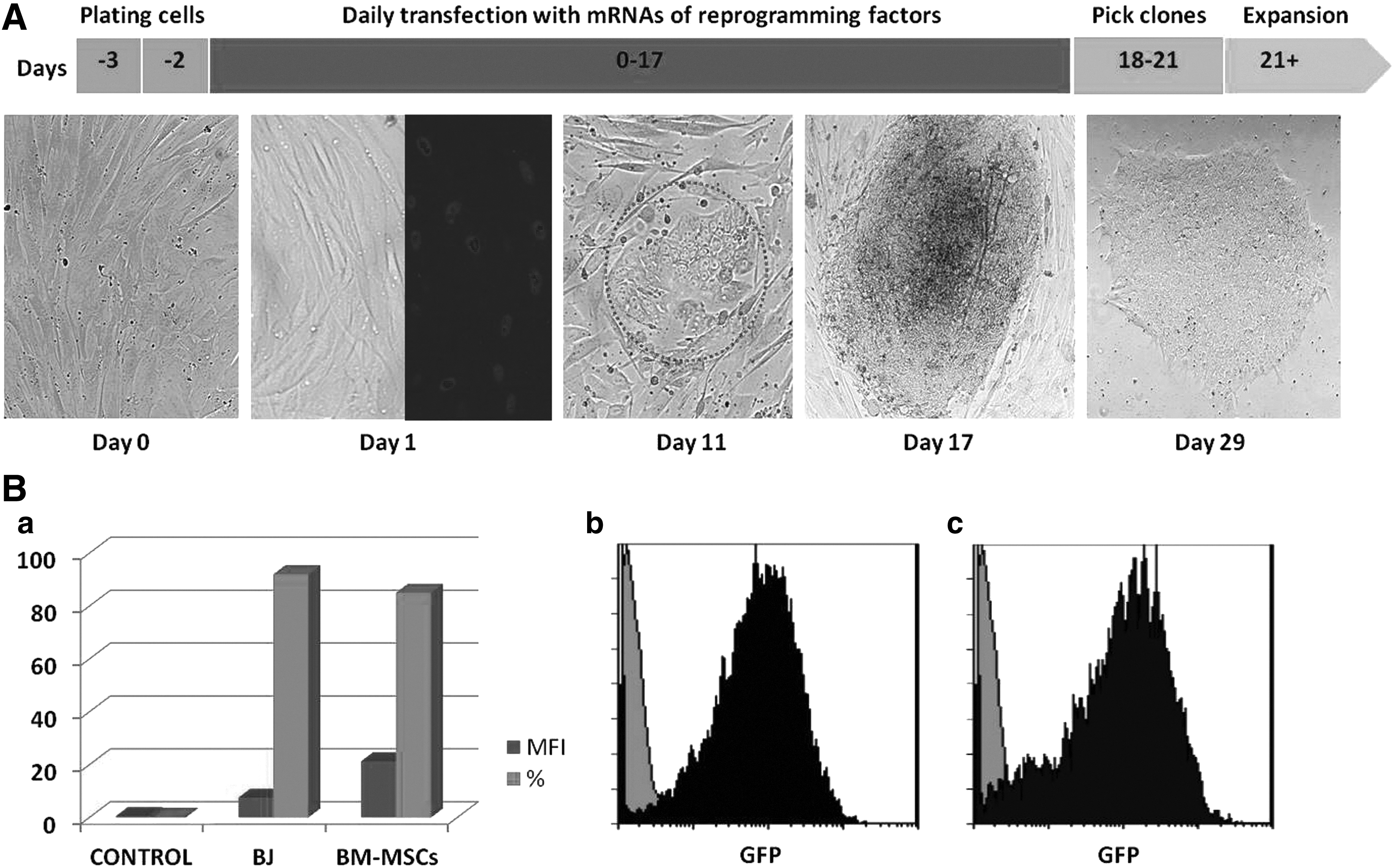
Transfection of human thalassemia BM-MSCs with modified mRNA. (
Phenotypic and molecular characterization of β-thal-iPSCs
A number of assays have been applied to detect pluripotent markers of the β-thal-iPSC lines that have been generated from BM-MSCs. All analyses were performed on samples obtained from passage (P) 10. Flow cytometry analysis revealed that β-thal-iPSC lines express high levels of the intracellular antigen Oct3/4 and the surface antigen SSEA-4 (Fig. 2A). RT-PCR results showed that β-thal-iPSCs express the pluripotency markers OCT3/4, REX-1, SOX2, and NANOG at levels equivalent to HUES-9 ESCs, whereas the original BM-MSCs had only low expression of OCT3/4 and NANOG (Fig. 2B). To confirm that the generated iPSCs were derived from the patient's cells, we confirmed the β-thal genotype for both BM-MSCs and derived iPSCs. When global gene expression was compared using Agilent microarrays analyzing the totality of known human transcripts, iPSC clones showed similar pattern to HUES-9 cells but not to parental BM-MSCs. Specifically, pluripotency-associated transcripts were clearly upregulated in the iPSCs compared to the parental BM-MSCs to levels comparable to hESCs (Fig. 2C).
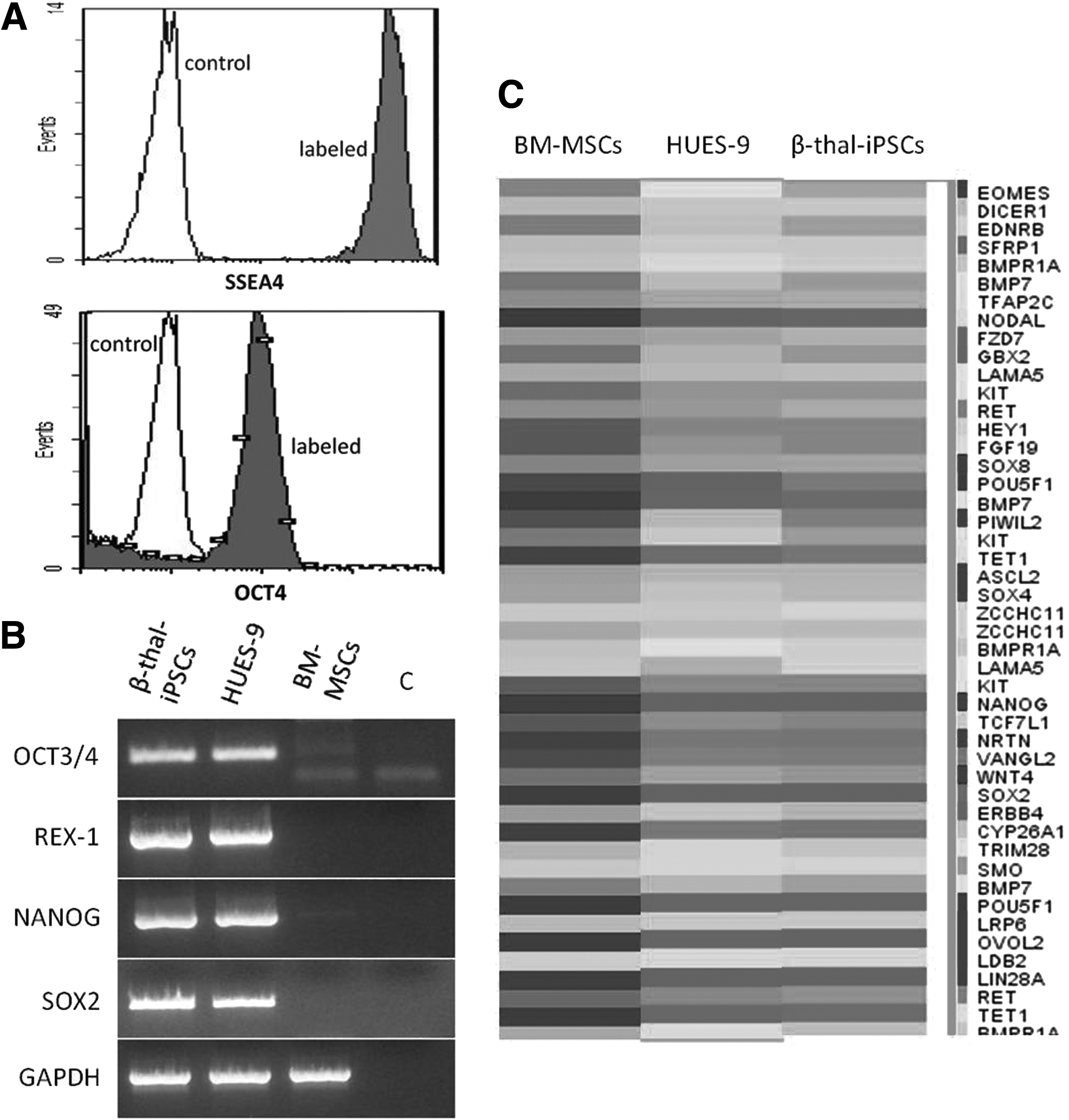
Expression of pluripotency markers in β-thal-iPSCs. (
To ensure that the reprogramming process did not result in any genetic alteration in the cells, we analyzed samples before the first passage of iPSCs with DNA array CGH (aCGH). We performed the same analysis at P10, to determine whether culture adaptation caused chromosomal aberrations in cells. Molecular karyotypes of iPSC lines were normal, and no genetic alteration was revealed in comparison to the original BM-MSCs (Fig. 3A).
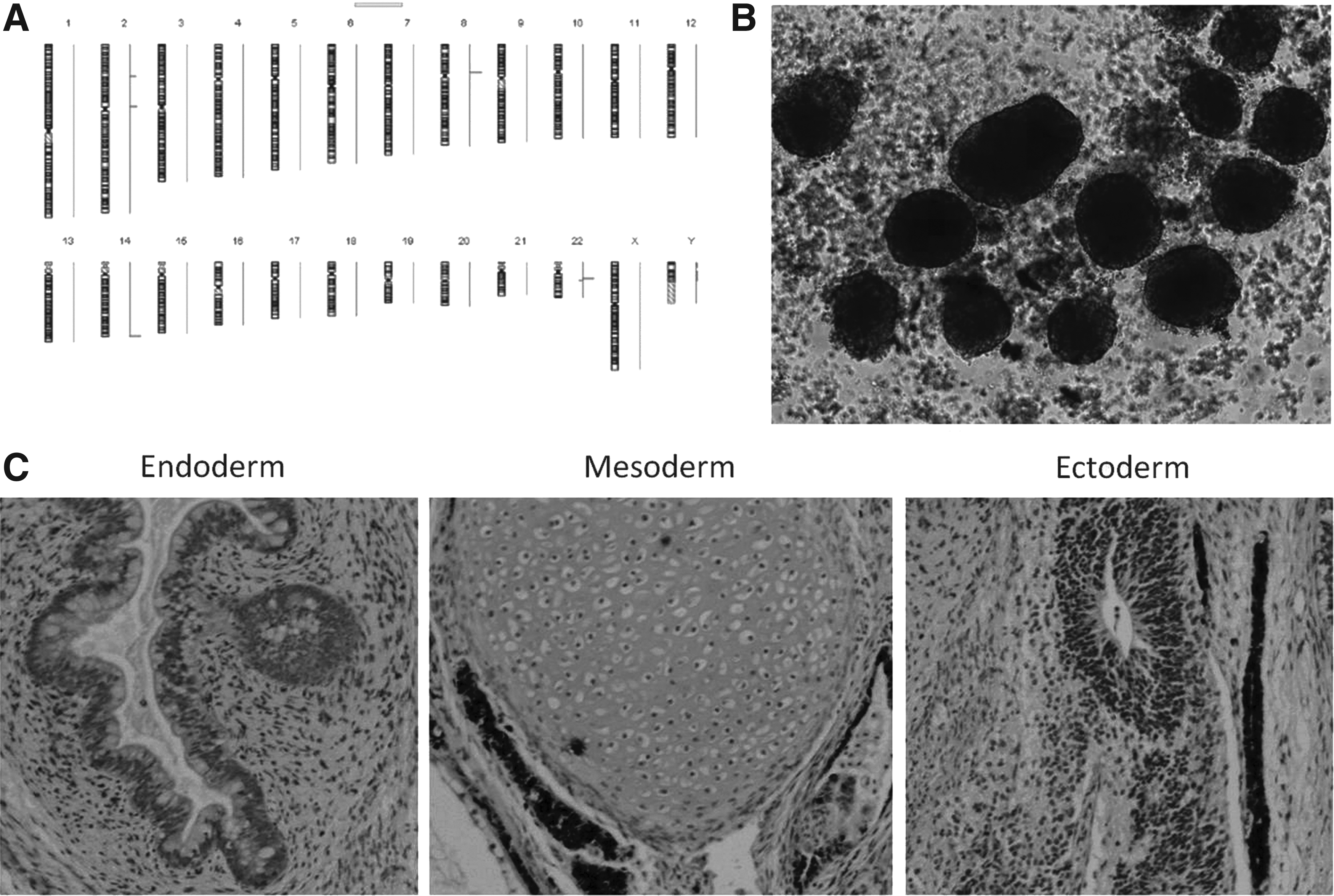
Genetic stability and differentiation potential of β-thal-iPSCs. (
In vitro differentiation and in vivo teratoma formation
Pluripotency was further confirmed both in vitro and in vivo by the formation of EBs and development of teratomas in SCID mice, respectively. After 7 days of culture in low-attachment plates, iPSCs formed EBs, indicating successful differentiation into the three germ layers in vitro (Fig. 3B). When β-thal-iPSCs were injected subcutaneously into immunodeficient mice, we observed teratoma formation after 7–11 weeks. Histological examination revealed that the teratomas contained tissues derived from all three germ layers, including intestinal epithelium (endoderm), cartilage (mesoderm), and neuroepithelium (ectoderm) (Fig. 3C).
In vitro hematopoietic differentiation
To investigate the ability of the β-thal-iPSCs to differentiate into hematopoietic progenitor cells, we used a feeder-free protocol based on the animal product–free medium STEMdiff APEL supplemented with growth factors, as detailed in Materials and Methods, in adherent cell culture, on a Matrigel-coated surface. For the experiments, iPSCs were used at P10. By day 8, we observed the formation of areas consisting of single small round cells on adherent cells (Fig. 4A). On day 13, cells were harvested and analyzed by flow cytometry. CD34 expression was detected up to 10% of the total cells, of which the majority revealed a dim CD45 co-expression, whereas only a minor cell population showed CD45 bright expression (Fig. 4B). Their hematopoietic potential was further examined by methylcellulose cloning assays. We observed a reduced clonogenic potential with the development of three to five granulocyte-monocyte colonies (GM-CFU)/105 cells, whereas erythroid colonies could not be detected. In contrast, using the same culture protocol, HUES-9 cells differentiated efficiently into CD34+CD45+ hematopoietic progenitors displaying normal colony-forming activity for both erythroid and granulomonocytic lineages.
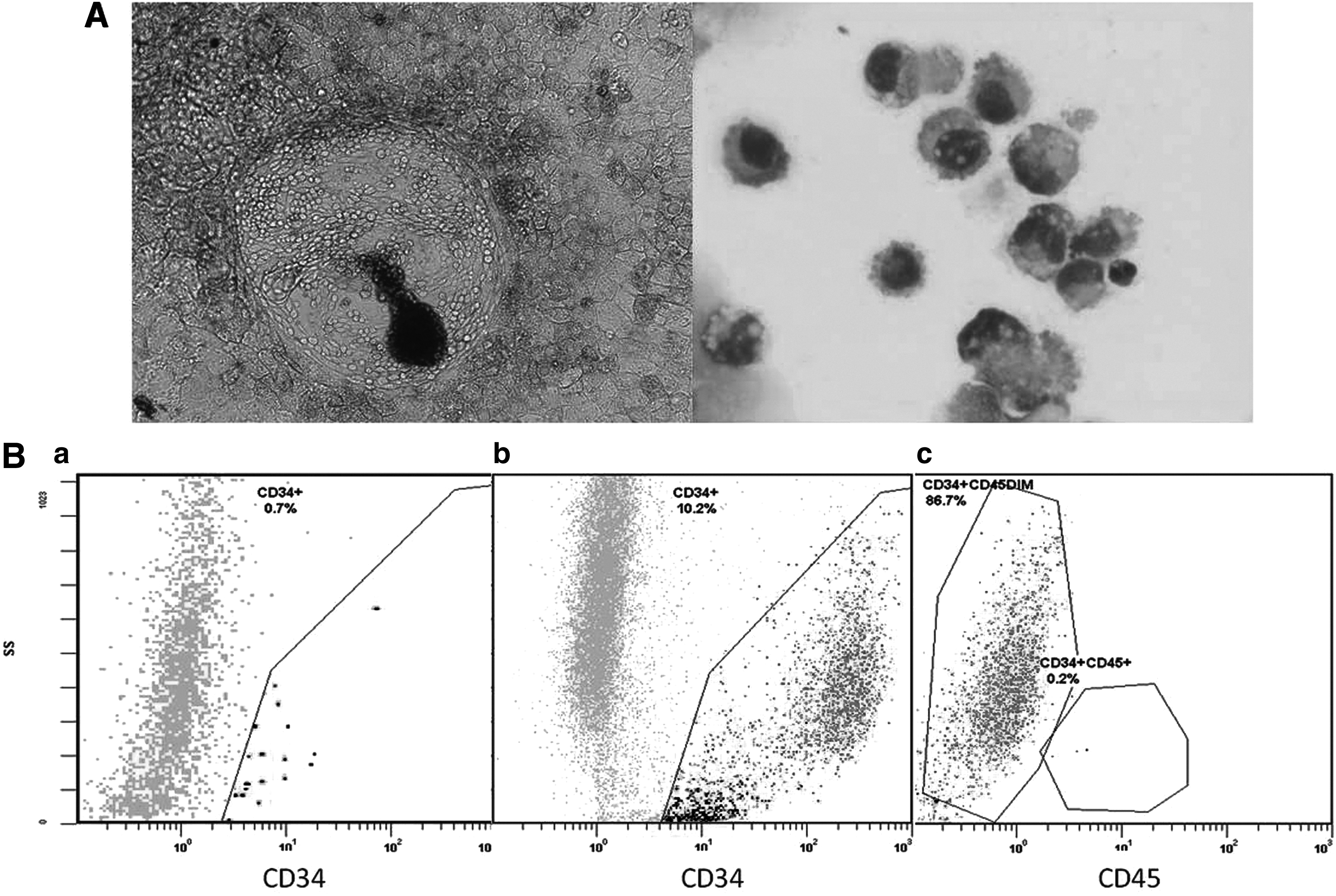
In vitro hematopoietic differentiation of β-thal-iPSCs. (
Discussion
With the aim of generating β-thal-iPSC lines that would be as safe as possible and thus applicable to cell therapy protocols, we chose to use a commercially available mRNA reprogramming protocol for three reasons. First, the host genome integrity is secured by the fact that the method does not involve the use of exogenous recombinant DNA constructs. Second, the method does not cause permanent or prolonged ectopic expression of the transcription factors, and third, it seems to be substantially simpler, faster, and potentially more efficient than most other technologies currently used.
We hypothesized that BM-MSCs can be reprogrammed efficiently to a pluripotency stage using a protocol with modified mRNA molecules originally described for fibroblasts. Both cell types have been grown in vitro as adherent cells exhibiting similar proliferative and differentiative potential (Haniffa et al., 2009), whereas BM-MSCs have been shown to be reprogrammed to iPSCs by using other technologies (Ohnishi et al., 2012). BM samples were available from autologous backup BM grafts harvested from thalassemic patients who were candidates for allogeneic BM transplantation in our stem cell transplant unit. Our results show that iPSC colonies were generated at day 15 after transfection of BM-MSCs with a reprogramming efficiency of 0.05%, which is about 74 times lower than the control cells (BJ fibroblasts).
Contradictory results have been reported on the reprogramming efficiency of mesenchymal cells using viral transfection methods, with ranges varying between 5% and 0.0002% (de Carvalho Rodrigues et al., 2012; Ohnishi et al., 2012). However, a recent report on reprogramming of adipose tissue–derived MSCs by a feeder-free mRNA method showed a reprogramming efficiency of 0.005% (Heng et al., 2013). We demonstrated that all β-thal-iPSC lines are pluripotent because they express markers of pluripotency, can differentiate in vitro to EBs, and form teratomas comprising tissues derived from all three germ layers when injected into immunodeficient mice. In addition, they do not exhibit genetic alterations after 10 culture passages in a Matrigel-based feeder-free medium as shown by aCGH, and they remain stable in maintaining pluripotency.
Both embryonic and iPSCs represent ideal cell populations for in situ correction of the disease-causing mutations by gene targeting methods. Using gene targeting mediated by homologous recombination or the recent developed transcription activator-like effector nucleases (TALEN) technology, gene-corrected iPSC lines have been generated (Ma et al., 2013; Papapetrou et al., 2011; Wang et al., 2012). Hemoglobin analysis of erythroblasts derived from corrected iPSC lines showed restoration of the expression of full-length β-globin. When hematopoietic progenitors, generated from genetically corrected β-thal-iPSCs by homologous recombination, were given to sublethally irradiated mice, an improved hemoglobin production was observed compared with infusion of uncorrected cells (Wang et al., 2012). However, further studies are needed to examine whether hematopoietic stem cells differentiated from corrected iPSCs can restore hematopoiesis in vivo.
The differentiation potential of human iPSC lines into hematopoietic cells has been well studied, and there are many reports showing successful hematopoietic specific differentiation (Choi et al., 2009; Lengerke et al., 2009; Ye et al., 2009b). We could generate CD34+ progenitors from iPSCs cultured for 10 passages by using a well-established feeder-free differentiation culture protocol (Ng et al., 2008); however, most of them were CD45dim and demonstrated decreased hematopoietic colony-forming capability. Our findings are in line with a recent report demonstrating that hemangioblastic derivatives from human iPSC lines exhibit substantially reduced hematopoietic colony-forming capability in comparison to those derived from human ESC lines (Feng et al., 2010). The decreased hematopoietic differentiation capacity has been also observed and reported by others investigators in early-passage iPSCs originated from somatic cells other than hematopoietic cells (Kim et al., 2011; Polo et al., 2010).
Recent studies showed that during the initial culture passages iPSCs maintain a set of epigenetic and transcriptional markers of their somatic cell of origin, a phenomenon defined as epigenetic donor cell memory. Such a residual transcriptional and epigenetic memory may be in favor of an in vitro differentiation potential of iPSCs back into their tissue of origin rather than into other cell types (Bar-Nur et al., 2011; Kim et al., 2010). Remarkably, this tendency of early-passage iPSCs to differentiate toward the cell lineage of origin seems to be lost over time after extending culture passaging (>25 passages) (Vitaloni et al., 2014).
One major hurdle for efficient generation of iPSC-derived hematopoietic stem cells is that all differentiation protocols achieve hematopoietic differentiation, which is limited to the generation of CD34+ hematopoietic stem/progenitor cells without engraftment potential and mature cells. Moreover, these CD34+ cells exhibit limited growth and expansion capability, and hence are not suitable for any in vivo use. Nevertheless, the generation of hematopoietic stem cells or progenitors with a limited proliferative potential may not be an unsolved problem, because it appears that the in vitro self-renewal potential can be activated in differentiated cells without tumorigenic transformation by combined ectopic overexpression of cMyc and Klf4 (Aziz et al., 2009). More importantly, Doulatov et al. reported recently a new approach that combines directed differentiation with transcription-based reprogramming to convert lineage-restricted CD34+CD45+ myeloid precursors derived from human pluripotent stem cells into hematopoietic multilineage progenitors that can be expanded in vitro and engrafted in vivo (Doulatov et al., 2013). A detailed in vitro and in vivo functional characterization of the iPSC-derived CD34+ hematopoietic stem/progenitor cells, including comparison with their normal BM counterparts so as to demonstrate their repopulating potential, must be an absolute requirement for each iPSC line before any possible clinical use.
In this study we have shown that reprogramming of BM-MSCs to pluripotent stem cells is feasible by applying the same synthetic mRNA protocol originally optimized for fibroblasts. Using the above methodology, we were able to generate three unequivocally safe β-thal-iPSC lines suitable for use in regenerative medicine.
Footnotes
Acknowledgments
We are grateful to Miltenyi Biotec, Bergisch Gladbach, Germany, for providing the reprogramming kit and continued support in our research program. We also thank Dr. K. Stefanaki for the histopathological assessment of teratoma tissue.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.