
Abstract
Abstract
Bovine somatic cell nuclear transfer (SCNT) using vitrified–thawed (VT) oocytes has been studied; however, the cloning efficiency of these oocytes is not comparable with that of nonvitrified (non-V) fresh oocytes. This study sought to optimize the survival and cryopreservation of VT oocytes for SCNT. Co-culture with feeder cells that had been preincubated for 15 h significantly improved the survival of VT oocytes and their in vitro developmental potential following SCNT in comparison to co-culture with feeder cells that had been preincubated for 2, 5, or 24 h (p<0.05). Spindle assessment via the Oosight Microscopy Imaging System and microtubule staining revealed that vitrified metaphase II oocytes (VT group) were not suitable for SCNT. However, enucleating and/or activating oocytes prior to freezing enhanced their developmental potential and suitability for SCNT. The cloning efficiency of the enucleated–activated–vitrified–thawed (EAVT) group (21.6%) was better than that of the other vitrification groups [enucleated–vitrified–thawed (EVT) group, 13.7%; VT group, 15.0%; p<0.05] and was comparable with that of the non-V group (25.9%). The reactive oxygen species level was significantly lower in the EAVT group than in the other vitrification groups (p<0.05). mRNA levels of maternal genes (ZAR1, BMP15, and NLRP5) and a stress gene (HSF1) were lower in the vitrification groups than in the non-V group (p<0.05), whereas the level of phospho-p44/42 mitogen-activated protein kinase did not differ among the groups. Among the vitrification groups, blastocysts in the EAVT group had the best developmental potential, as judged by their high mRNA expression of developmental potential–related genes (POU5f1, Interferon-tau, and SLC2A5) and their low expression of proapoptotic (CASP3) and stress (Hsp70) genes. This study demonstrates that SCNT using bovine frozen–thawed oocytes can be successfully achieved using optimized vitrification and co-culture techniques.
Introduction
S
Vitrification (glass-like solidification) reduces oocyte damage during cryopreservation by increasing the cooling and warming rates, thereby avoiding ice crystal formation (Rall and Fahy, 1985). The oocytes of many mammalian species have been successfully vitrified, including those of cattle (Martino et al., 1996), mice (Men et al., 1997), pigs (Isachenko et al., 1998), and humans (Kuwayama et al., 2005). Many simple and efficient vitrification methods for bovine oocyte cryopreservation have been reported. These use various containers (e.g., electron microscopy grid, open pulled straw, Cryoloop, and solid surface) and a small volume (1–2 μL) of freezing solution, which has a high cooling capacity (Dinnyés et al., 2000; Lane et al., 1999; Martino et al., 1996; Vajta et al., 1998). Nevertheless, it remains difficult to cryopreserve bovine oocytes, and the developmental potential and cloning efficiency of vitrified–thawed (VT) oocytes following SCNT are poor (Booth et al., 1999; Hou et al., 2005; Yang et al., 2008).
To enhance the cloning efficiency of SCNT using cryopreserved bovine oocytes, it might be necessary to minimize freezing, mechanical, and chemical damage such that all frozen–thawed oocytes survive. Co-culture with somatic cells can be used to overcome the developmental block of frozen–thawed oocytes (Fukuda et al., 1990; Bavister, 1992). In previous studies (Kim et al., 2012; Kim et al., 2013), we performed SCNT with ear cells and co-cultured the resultant embryos with the same cells. Optimized feeder cell conditions may be key to improving the survival and developmental potential of oocytes after thawing. Another concern is that microtubule depolymerization induced by CPA treatment and cryopreservation can cause meiotic spindle disassembly and chromosome misalignment (Shi et al., 2006); therefore, the results of SCNT studies using VT metaphase II (MII) oocytes might be highly variable and not comparable with those of SCNT studies using fresh oocytes (Booth et al., 1999; Hou et al., 2005; Muenthaisong et al., 2007; Yang et al., 2008). In our previous study (Kim et al., 2001), we successfully vitrified bovine oocytes using the minimum volume cooling (MVC) method and confirmed that these frozen–thawed oocytes can develop to full-term following in vitro fertilization (IVF). However, the use of the MVC method for SCNT was not optimized (Park et al., 2002).
The current study sought to optimize conditions for the survival and cryopreservation of VT oocytes used for SCNT. Changes in the reactive oxygen species (ROS) level, expression of maternal-related genes [zygote arrest 1 (ZAR1), bone morphogenenic protein 15 (BMP15), growth differentiation factor 9 (GDF9), NLR family, pyrin domain-containing 5 (NLRP5), heat shock transcription factor 1 (HSF1), and superoxide dismutase 1 (SOD1)], and mitogen-activated protein kinase (MAPK) activity may be useful for analyzing the viability of frozen–thawed oocytes used for SCNT and the freezing-related decline in the survival of these oocytes.
In the current study, we show that SCNT can be performed using VT oocytes, as well as nonvitrified (non-V) fresh oocytes. We examined the following: (1) Optimization of the survival of VT oocytes used for SCNT; (2) how the cloning efficiency is affected by changes to spindle integrity following freezing and thawing; and (3) comparison of the cloning efficiency among oocytes that are conventionally vitrified, those that are enucleated prior to freezing, and those that are enucleated and activated prior to freezing. We also compared these various VT oocytes and fresh oocytes in terms of the level of ROS, microfilament localization, mRNA expression of maternal candidate genes (ZAR1, BMP15, GDF9, NLRP5, HSF1, and SOD1), and the level of phosphorylated MAPK. The mRNA levels of developmental potential–related genes [POU class 5 homeobox 1 (POU5f1), Interferon-tau, solute carrier family2 (facilitated glucose/fructose transporter) member5 (SLC2A5), caspase 3 (CASP3), heat shock protein 70 (Hsp70), and DNA (cytosine-5)-methyltransferase 3-alpha (Dnmt3A)] were compared among blastocysts produced from the various groups of oocytes.
Materials and Methods
Unless stated otherwise, all chemicals were purchased from Sigma Chemical Company (St. Louis, MO, USA).
Oocyte preparation and in vitro maturation
Bovine ovaries were collected from a slaughterhouse and transported to the laboratory within 2 h in 0.9% saline solution at 35°C. Cumulus–oocyte complexes (COCs) were aspirated from visible follicles (2–6 mm) with an 18-gauge needle attached to a 10-mL disposable syringe. The medium used for COC collection was HEPES-buffered Tyrode's medium (TL-HEPES). Sets of ten COCs were matured in vitro in tissue culture medium-199 (TCM-199; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 0.2 mM sodium pyruvate, 1 μg/mL follicle-stimulating hormone (Folltropin™, Bioniche Animal Health, Belleville, ON, Canada), 1 μg/mL estradiol-17β, and 1 mM epidermal growth factor (EGF) in mineral oil at 38.8°C in an incubator (5% CO2, 5% O2, and 90% N2) for 19–21 h.
Vitrification and thawing
The basic medium used for pretreatment, vitrification, and dilution was Dulbecco's phosphate-buffered saline (D-PBS, Gibco) containing 10% FBS. The pretreatment solution also contained 10% ethylene glycol (EG10). The vitrification solution (VS) contained 30% ethylene glycol and 0.5 M sucrose (EG30). For serial dilution after thawing, D-PBS containing 1.0, 0.5, 0.25, or 0.125 M sucrose and 10% FBS was used. Oocytes were freeze–thawed according to the MVC vitrification procedures reported previously (Kim et al., 2001).
After incubation for 20 h in In Vitro Maturation (IVM) medium, cumulus cells were partially (MII oocytes) or completely (enucleated oocytes) removed by treatment with 0.1% hyaluronidase and mechanical pipetting. Oocytes were washed with TL-HEPES, incubated in a droplet of previous cultured IVM medium for 1 h to recover, and then frozen with or without prior enucleation and/or activation. Freezing procedures were performed at room temperature. MII oocytes or enucleated oocytes were washed three times in TL-HEPES and then equilibrated in D-PBS for 5 min. For vitrification, oocytes were pretreated with EG10 for 5 min, exposed to EG30 for 30 sec, and then loaded individually onto the inner wall of a modified French ministraw (total length, 2.5–3.0 cm) coated with a minimum volume of VS. The straw was plunged directly into liquid N2, and four to five straws were placed into a prechilled cryovial, which was stored in a freezing cane and placed in a liquid nitrogen tank.
For thawing, CPAs were removed via a five-step procedure using thawing solutions warmed to 37°C. Straws stored in liquid nitrogen were moved rapidly to D-PBS containing 1.0 M sucrose. Thereafter, oocytes were sequentially transferred to D-PBS containing 0.5, 0.25, and 0.125 M sucrose, and then into D-PBS lacking sucrose. Oocytes were incubated in each solution for 1 min. Finally, oocytes were cultured with feeder cells (preincubated for 2, 5, 15, or 24 h) in TCM-199 medium for 2 h.
Preparation of donor cells and feeder cells
Donor somatic cells were derived from the ear tissue of Hanwoo Cattle (Korean Native Cattle). Minced ear tissue was incubated in 0.1% collagenase type IV solution at 38°C for 1.5 h and then cultured in donor cell culture medium [Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 1 mM sodium pyruvate, 1% nonessential amino acids, 0.1% β-mercaptoethanol, and 1% penicillin-streptomycin]. The cells were grown and subcultured three to five times, with an interval of 4–6 days. Thereafter, cells (1×106) were frozen in cryovials (1.5 mL) in freezing medium (50% donor cell culture medium containing 45% FBS and 5% dimethyl sulfoxide). For SCNT, frozen–thawed ear cells were washed twice with donor cell culture medium and treated with 3 mg/mL protease for 50 sec at room temperature. Treated cells were washed three times and resuspended in donor cell preparation medium (TCM-199 HEPES supplemented with 0.2 mM sodium pyruvate).
A droplet of feeder cells was prepared using cultured bovine ear cells, the same cells as were used for SCNT, to create homogeneous culture conditions for oocytes and embryos. The cells were detached using TrypLE reagent (Gibco), added to PBS, centrifuged at 2000×g for 1 min, resuspended in DMEM containing 10% FBS, and seeded into a 10-μL droplet. The droplet was covered with mineral oil and incubated at 38.8°C in 5% O2, 5% CO2, and 90% nitrogen for 1 or 2 days prior to co-culture with frozen–thawed oocytes.
Preparation of recipient oocytes
For enucleation, cumulus cells were completely removed from the oocyte by vortexing for 3 min in the presence of 0.05% hyaluronidase. Oocytes with an extruded first polar body (PB1) were selected, and denuded oocytes were transferred to enucleation medium (TCM-199 HEPES containing 20% FBS and 7.5 μg/mL cytochalasin B). Thereafter, the MII plate and PB1 were visualized using an inverted microscope (Olympus, Tokyo, Japan) equipped with the Oosight Microscopy Imaging System (CRi, Hopkinton, MA, USA) and removed by the squeezing method, as reported previously (Kim et al., 2012).
Somatic cell nuclear transfer
A single treated donor cell was placed in the perivitelline space of an enucleated oocyte in nuclear transfer medium [TCM-199 HEPES containing 0.06% fatty acid–free bovine serum albumin (BSA) and 10 μg/mL phytohemagglutinin] through the opening made during enucleation. Thereafter, oocytes were placed in cell fusion medium (0.3 M mannitol, 0.5 mM HEPES, 0.05 mM CaCl2, and 0.1 mM MgSO4) and subjected to an electrical pulse of 1.3 kV/cm for 20 μsec using an Electro Cell Fusion Generator (LF101, NEPAGENE, Chiba, Japan). After fusion, the reconstructed embryos were kept in TCM-199 HEPES supplemented with 20% FBS for 1 h, activated for 5 min in CR1aa medium supplemented with 1.5 mg/mL BSA and 10 μM calcium ionophore, and exposed to 2 mM 6-dimethylaminopurine for 3 h. The reconstructed embryos were cultured in CR1aa medium supplemented with 10% FBS, 1 μM EGF, 1 μM insulin-like growth factor, and 10 μM flavonoid at 38.8°C in 5% O2, 5% CO2, and 90% nitrogen with maximum humidity for 8 days.
Immunofluorescence microscopy
Microtubules and DNA were detected by indirect immunocytochemical techniques, as described by Kim et al. (1996). Briefly, oocytes were permeabilized in modified Buffer M (25% glycerol, 50 mM KCl, 0.5 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid, 1 mM β-mercaptoethanol, 50 mM imidazole, pH 6.7, 3% Triton X-100, and 25 mM phenylmethylsulfonyl fluoride; Simerly and Schatten, 1993) for 20 min, fixed in methanol at −20°C for 10 min, and stored in PBS containing 0.02% sodium azide for 7 days at 4°C. Microtubule localization was determined using an anti-α-tubulin antibody (T-5168). Fixed oocytes were incubated for 90 min at 39°C with this antibody diluted 1:100 in PBS. After several washes with PBS containing 0.5% Triton X-100 and 0.5% BSA, oocytes were incubated in blocking solution (0.1 M glycine, 1% goat serum, 0.01% Triton X-100, 1% powdered milk, 0.5% BSA, and 0.02% sodium azide) at 38°C for 1 h. Thereafter, oocytes were incubated with a fluorescein isothiocyanate (FITC)-labeled goat anti-mouse antibody. DNA was detected fluorescently by incubation with 50 μg/mL propidium iodide for 1 h. Following extensive washing, samples were mounted onto glass slides. Fluorescence images of individual samples were digitized using a laser scanning confocal microscope (Leica Laser Technik GmbH) and MetaMorph software (v. 6.1, Universal Imaging Corporation, Westchester, PA, USA).
To detect the distribution of microfilaments, oocytes were fixed with 4% paraformaldehyde prepared in PBS for 1 h and then transferred to membrane permeabilization solution (0.5% Triton X-100) for 30 min. After 1 h in blocking buffer (1% BSA prepared in PBS), oocytes were incubated with phalloidin-tetramethylrhodamine (TRITC) (1 μg/mL) for 2 h. After three washes (5 min each) with 1% BSA prepared in PBS, samples were co-stained with Hoechst 33342 (1 μg/mL prepared in PBS) for 30 min. Oocytes were mounted onto glass slides and examined using a confocal laser scanning microscope (Leica Laser Technik GmbH). At least 20 oocytes were examined per group.
Measurement of intracellular ROS
Intracellular ROS were measured in oocytes by a 2,7-dichlorofluorescein assay, as described previously (Lee et al., 2011). Briefly, oocytes were incubated with 100 μM 2,7-dichlorodihydrofluorescein diacetate (Molecular Probes, Eugene, OR, USA) for 20 min at 38°C, washed three times in TL-HEPES to remove excess dye, and immediately analyzed by epifluorescence microscopy (Olympus, Tokyo, Japan) using excitation and emission wavelengths of 450–490 nm and 515–565 nm, respectively. Grayscale images were acquired using a digital camera (Nikon, Tokyo, Japan) attached to the microscope, and mean gray values were measured using ImageJ (NIH, Bethesda, MD, USA). Background fluorescence values were subtracted from the final values prior to statistical analysis. Experiments were repeated three times, with 10–20 oocytes per experiment.
Real-time reverse transcription PCR
Comparative real-time reverse transcription (RT)-PCR was performed using a Chromo 4 detector (Bio-Rad) and a DyNAmo HS SYBR Green qPCR Kit (FINNZYMES), according to the manufacturer's instructions. The primers are described in Table 1. Magnetic beads (Dynabeads mRNA Purification Kit; Dynal, Oslo, Norway) were used to prepare mRNA from 20 oocytes per group. Gene expression was quantified by the 2−ΔΔCt method (Livakand Schmittgen, 2001).
bZAR1, bovine zygote arrest 1; bBMP15, bone morphogenetic protein 15; bGDF-9, bovine growth differentiation factor 9; bNLRP5, bovine NLR family, pyrin domain-containing 5; bHSF1, bovine heat shock transcription factor 1; bSOD1, superoxide dismutase 1; bPOU5f1, bovine POU class 5 homeobox 1; bInterferon-tau, bovine Interferon-tau; bSLC2A5, bovine solute carrier family 2 (facilitated glucose/fructose transporter) member 5; bCASP3, bovine caspase 3; bHsp70, bovine heat-shock protein 70; bDnmt3A, bovine DNA (cytosine-5)-methyltransferase 3-alpha; bGAPDH, bovine glyceraldehyde 3-phosphate dehydrogenase.
F, forward; R, reverse.
Western blot analysis
Oocytes (30 per sample) were added to 20 μL of 1×sodium dodecyl sulfate (SDS) sample buffer [62.5 mM Tris-HCl (pH 6.8 at 25°C), 2% (wt/vol) SDS, 10% (vol/vol) glycerol, 50 mM dithiothreitol (DTT), and 0.01% (wt/vol) Bromophenol Blue or Phenol Red] and heated for 5 min at 95°C. Proteins were resolved on a 5–12% Tris-SDS-polyacrylamide gel electrophoresis (PAGE) gel for 1.5 h at 80–100 V. Thereafter, proteins were transferred to a nitrocellulose membrane (Amersham, Hybond-ECL, Buckinghamshire, UK) at 300 mA for 2 h in transfer buffer [25 mM Tris (pH 8.5) containing 200 mM glycine and 20% (vol/vol) methanol]. After blocking for 1 h with 5% skimmed milk prepared in PBS, the membrane was incubated for at least 2 h with anti-phospho-p44/42 MAPK and anti-MAPK antibodies (Cell Signaling Technology, Danvers, MA, USA) diluted 1:500 in blocking solution [1×Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% (wt/vol) nonfat dry milk], washed three times in TBST [20 mM Tris-HCl (pH 7.5) containing 250 mM NaCl and 0.1% (vol/vol) Tween-20], and incubated for 1 h with anti-rabbit immunoglobulin G horseradish peroxidase (IgG-HRP; Cell Signaling Technology, Danvers, MA, USA) diluted 1:2000 in blocking solution. After three washes with TBST, antibody binding was visualized with a chemiluminescence luminol reagent (Invitrogen, Carlsbad, CA, USA).
Experimental design
Oocytes were divided into the following groups—enucleated–vitrified–thawed (EVT), enucleated–activated–vitrified–thawed (EAVT), VT, and non-V. To determine the condition in which feeder cells best support the survival of VT bovine oocytes used for SCNT, feeder cells were preincubated for various amounts of time (2, 5, 15, and 24 h). The suitability of VT MII oocytes for SCNT was determined by assessing the spindle using the Oosight Microscopy Imaging System (live imaging) and immunofluorescence labeling of microtubules (fixed samples). The developmental potentials of oocytes in the EVT and EAVT groups were compared with those of oocytes in the non-V and VT groups. Furthermore, the levels of ROS, microfilament localization, and mRNA expression of maternal candidate genes (ZAR1, BMP15, GDF9, NLRP5, HSF1, and SOD1) and the level of phosphorylated MAPK in oocytes were compared among the groups. In addition, the mRNA levels of developmental potential–related genes (POU5f1, Interferon-tau, SLC2A5, CASP3, Hsp70, and Dnmt3A) in in vitro–produced 8-day-old SCNT blastocysts were compared among the various groups. Experiments were repeated three to six times.
Statistical analysis
The general linear model procedure embedded in the Statistical Analysis System (SAS User's Guide, 1985, Statistical Analysis System Inc., Cary, NC, USA) was used to analyze all data. Significant differences were determined by the Tukey multiple range test. A paired Student t-test was used to compare relative gene expression. p values <0.05 were considered significant.
Results
Effect of the preincubation time of feeder cells on the survival of VT bovine MII oocytes used for SCNT
To determine the feeder cell conditions that best support the survival of bovine frozen–thawed MII oocytes used for SCNT, 10-μL droplets of feeder cells were preincubated for 2, 5, 15, and 24 h, and were then co-cultured with the oocytes (Table 2). The survival rates of oocytes co-cultured with feeder cells that had been preincubated for 5 h or longer [5 h, 88.2% (82/93); 15 h, 90.2% (83/92); 24 h, 86.7% (78/90)] were significantly higher than those of oocytes co-cultured with feeder cells that had been preincubated for 2 h (73.9%, 65/88; p<0.05). Fewer than half (44.9–50.0%) of the surviving VT oocytes could be used for SCNT. The percentage of oocytes that underwent fusion following SCNT was significantly higher among oocytes co-cultured with feeder cells that had been preincubated for 5 or 15 h [5 h, 80.5% (33/41); 15 h, 85.0% (34/40)] than among oocytes co-cultured with feeder cells that had been preincubated for 2 h (66.7%, 20/30) (p<0.05).
Non-V, nonvitrified MII oocyte group. To support the survival of vitrified–thawed (VT) MII oocytes, feeder cells used for co-culture were preincubated for various amounts of time (2, 5, 15, and 24 h). Oocytes were co-cultured with feeder cells for 2 h.
p<0.05.
M, metaphase; SCNT, somatic cell nuclear transfer; VT, vitrified–thawed.
However, the percentage of fused oocytes that underwent cleavage was significantly higher among oocytes co-cultured with feeder cells that had been preincubated for 15 h or longer [15 h, 52.9% (18/34); 24 h, 44.0% (11/25)] than among oocytes co-cultured with feeder cells that had been preincubated for 5 h or less [2 h, 35.0% (7/20); 5 h, 36.4% (12/33)]. In comparison, the cleavage rate was significantly higher in the non-V group (p<0.05). The percentage of blastocysts was significantly higher among oocytes co-cultured with feeder cells that had been preincubated for 15 h or longer [15 h, 27.8% (5/18); 24 h, 18.2% (2/11)] than among oocytes co-cultured with feeder cells that had been preincubated for 5 h or less [2 h, 0% (0/7); 5 h, 0% (0/7)]. Preincubation of feeder cells for 15 h resulted in the highest percentage of fused VT oocytes developing to the blastocyst stage (14.7%, 5/34); for comparison, the corresponding percentage in the non-V group was 23.6% (13/55; p<0.05).
Assessment of VT MII oocytes in terms of their survival following thawing, spindle visualization using the Oosight Microscopy Imaging System, and microtubule immunostaining
VT MII oocytes were assessed in terms of survival following thawing, visualization of the spindle using the Oosight Microscopy Imaging System (live imaging), and immunofluorescence labeling of microtubules (fixed samples) (Fig. 1). After thawing, 91.4% of MII oocytes survived. Spindle poles and polar bodies were visualized using the Oosight Microscopy Imaging System (Fig. 1B). From this, 48.8% (42/86), 34.9% (30/86), and 16.3% (14/86) of oocytes were judged to have spindle poles and polar bodies that were clear, unclear, and could not be observed (negative), respectively. Oocytes with spindle poles and polar bodies that could be visualized (clear and unclear) were subjected to microtubule immunostaining and confocal microscopy (Fig. 1C). In normal spindles, chromosomes were clustered in a discrete bundle at the metaphase plate were scattered along microtubules. The percentage of oocytes with normal microtubule staining was significantly higher among oocytes whose spindle poles and polar bodies were clearly visualized using the Oosight Microscopy Imaging System (64.3%, 27/42) than among those whose spindle poles and polar bodies were unclear using this system (43.3%, 13/30; p<0.05).
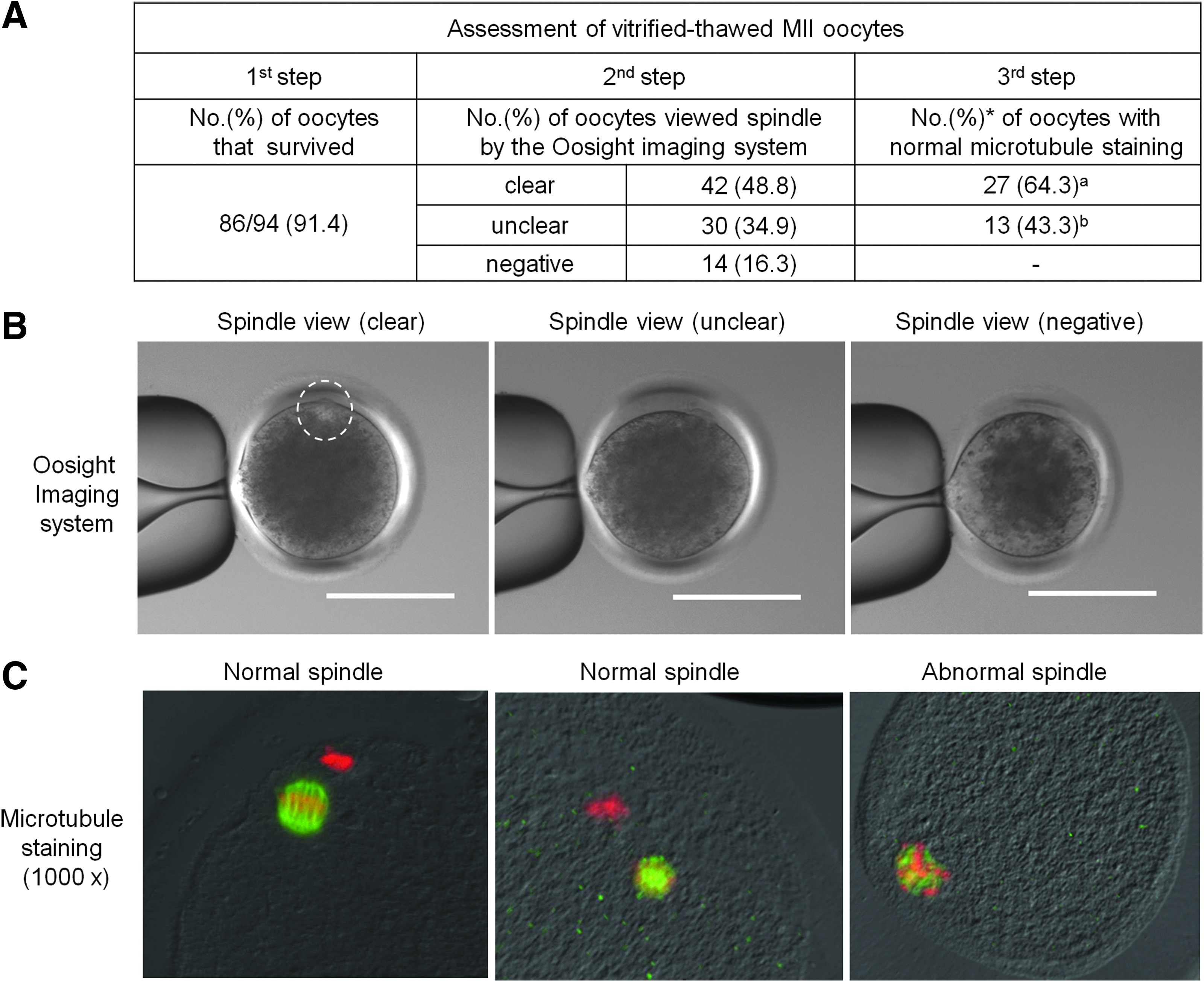
Vitrified–thawed bovine MII oocytes were assessed in terms of their survival following thawing, visualization of their spindle using the Oosight Microscopy Imaging System, and immunofluorescence labeling of microtubules. (
Effect of enucleation and activation prior to freezing on the cloning efficiency of bovine oocytes
Oocytes were enucleated (EVT group, Fig. 2E–G) or were enucleated and activated (EAVT group, Fig. 2I–K) prior to freezing. The developmental potentials of the resulting embryos (Fig. 2H, L), following thawing and SCNT, were compared with those of the non-V (Fig. 2M, N) and VT (Fig. 2A–D) groups. As shown in Table 3, the percentage of thawed oocytes that survived was similar among the VT (93.4%, 170/182), EVT (86.7%, 156/180), and EAVT (88.9%, 160/180) groups, although the percentage of oocytes that were suitable for SCNT significantly differed [VT, 52.9% (90/170); EVT, 78.2% (122/156); EAVT, 81.3% (130/160); p<0.05]. The percentage of vitrified oocytes that underwent fusion following SCNT [VT, 88.9% (80/90); EVT, 83.6% (102/122); EAVT, 89.2% (116/130)] was similar to the corresponding percentage in the non-V group [90.6% (116/128)].

Morphology of oocytes in the various groups, namely, vitrified–thawed (VT;
Non-V, nonvitrified MII oocyte group; VT, vitrified–thawed MII oocyte group; EVT, enucleated–vitrified–thawed oocyte group; EAVT, enucleated–activated–vitrified–thawed oocyte group. Oocytes were co-cultured for 2 h with feeder cells that had been preincubated for 15 h.
p<0.05.
SCNT, somatic cell nuclear transfer.
At day 2 postactivation, the percentage of embryos that underwent cleavage in the EAVT group [70.7% (82/116)] was significantly higher than that in the other vitrification groups [VT, 52.5% (42/80); EVT, 57.8% (59/102); p<0.05], but was not significantly different to that in the non-V group [72.4% (84/116)]. At day 8 postactivation, the percentage of embryos that had reached the blastocyst stage was higher in the EAVT group [30.5% (25/82)] than in the VT [28.6% (12/42)] and EVT [23.7% (14/59)] groups; the corresponding percentage in the non-V group was 35.7% (30/84). Finally, the overall efficiency with which SCNT embryos were produced in the EAVT group [21.6% (25/116)] was higher than that in the other vitrification groups [VT, 15.0% (12/80); EVT, 13.7% (14/102); p<0.05] and did not differ from that in the non-V group [25.9% (30/116)]. Therefore, among the vitrification groups, the cloning efficiency was highest in the EAVT group, with two-fold more in vitro–produced blastocysts (25) in this group than in the VT group (12).
Comparison of the levels of ROS and phosphorylated MAPK, microfilament organization, and mRNA expression among oocytes in the various groups
Oocytes in the VT, EVT, and EAVT groups were compared with those in the non-V group in terms of the level of ROS, microfilament localization, mRNA expression of maternal candidate genes (ZAR1, BMP15, GDF9, NLRP5, HSF1, and SOD1), and the level of phosphorylated MAPK (Fig. 3). The level of ROS in the EAVT group was significantly lower (Fig. 3A, d) than that in the VT (Fig. 3A, b) and EVT (Fig. 3A, c) groups, but was similar to that in the non-V group (Fig. 3A, a) (Fig. 3B, p<0.05). Microfilament localization did not differ among the various treatment groups, apart from the absence of spindle and chromatin labeling in the EVT (Fig. 3A, g) and EAVT (Fig. 3A, h) groups. mRNA levels of maternal genes (ZAR1, BMP15, and NLRP5) and the stress gene HSF1 were lower in the VT, EVT, and EAVT groups than in the non-V group (p<0.05). mRNA expression of the GDF9 (maternal) gene was significantly lower in the EVT group than in the non-V group, whereas mRNA expression of the SOD1 (antioxidant) gene was higher in the vitrification groups than in the non-V group (Fig. 3C). The level of phosphorylated MAPK did not vary among the groups (Fig. 3D).
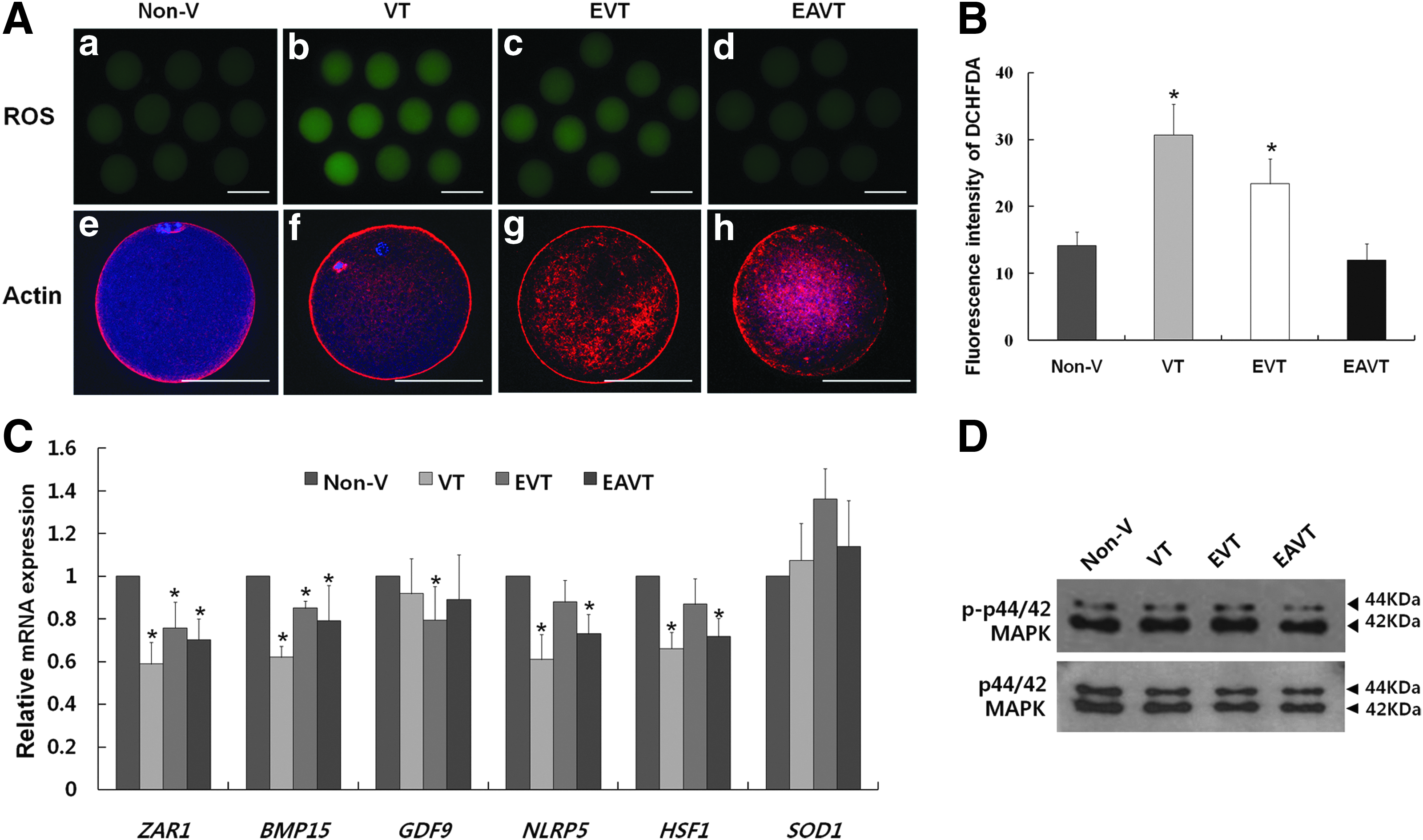
Levels of ROS, mRNA expression, and phosphorylated MAPK in oocytes in the various groups, namely, vitrified–thawed (VT), enucleated–vitrified–thawed (EVT), and enucleated–activated–vitrified–thawed (EAVT), were compared with those in the nonvitrified (non-V) group. (
Comparison of blastocyst mRNA expression of developmental potential–related genes among the groups
The mRNA levels of developmental potential–related genes (POU5f1, Interferon-tau, SLC2A5, CASP3, Hsp70, and Dnmt3A) in in vitro–produced 8-day-old SCNT blastocysts were compared among the groups. The mRNA levels of POU5f1 (pluripotency) and Interferon-tau (implantation) in these blastocysts were significantly lower in the EVT, VT, and EAVT groups (EVT<VT<EAVT) than in the non-V group (p<0.05; Fig. 4). The mRNA level of SLC2A5 (metabolism) did not differ between the non-V and EAVT groups, but was significantly lower in the VT and EVT groups (p<0.05). The mRNA levels of CASP3 (proapoptotic) and Hsp70 (stress) were significantly lower in the EVT and EAVT groups than in the non-V and VT groups (p<0.05), whereas the mRNA level of Dnmt3A (methylation) was significantly lower in the EVT group than in the non-V group.
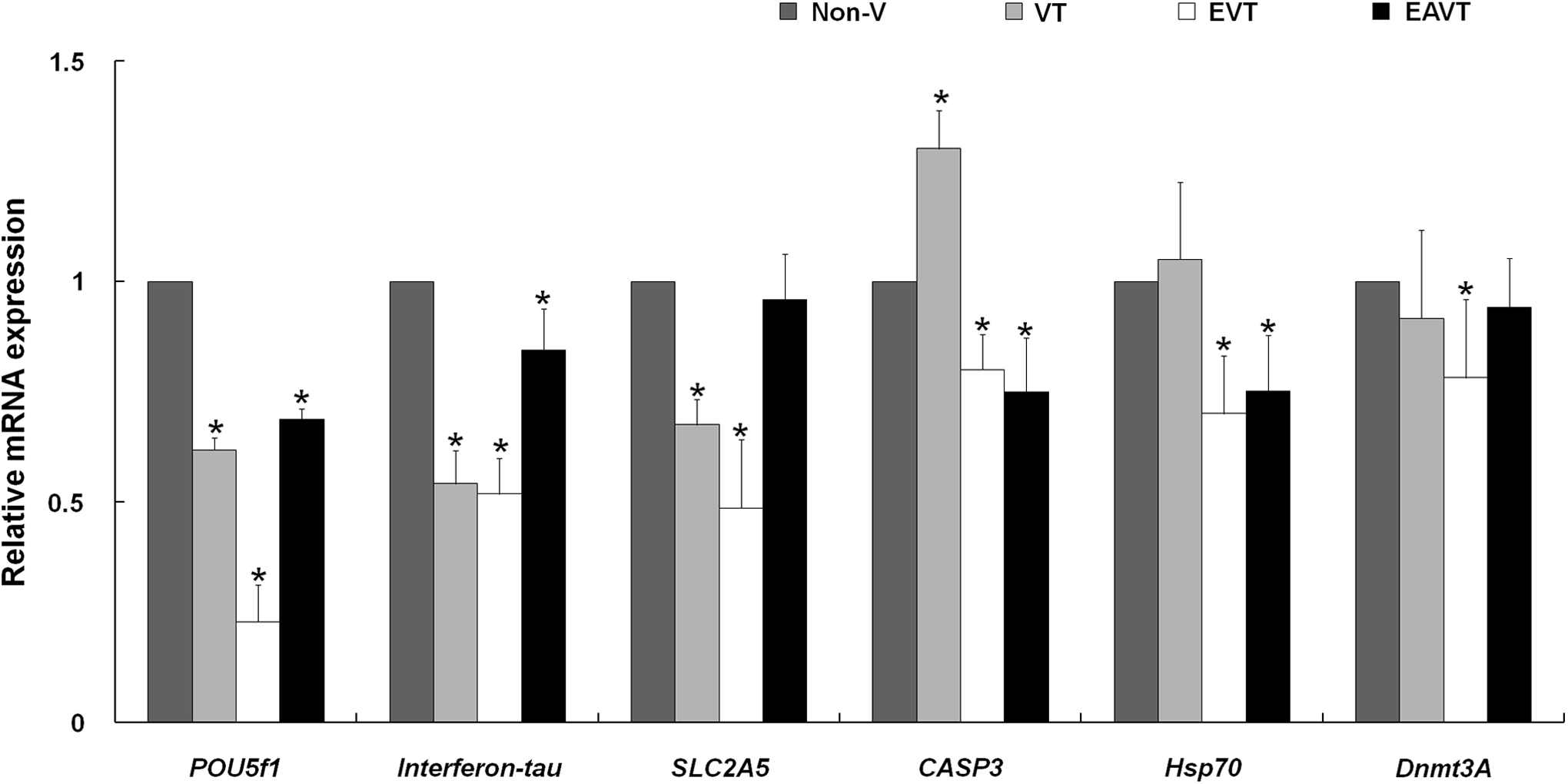
mRNA expression of developmental potential–related genes (POU5F1, Interferon-tau, SLC2A5, CASP3, HSP70, and Dnmt3A) in blastocysts produced via somatic cell nuclear transfer (SCNT) in the nonvitrified (non-V), vitrified–thawed (VT), enucleated–vitrified–thawed (EVT), and enucleated–activated–vitrified–thawed (EAVT) groups. GAPDH was used as an internal standard. (*) p<0.05 compared with the non-V group.
Discussion
This study demonstrates that SCNT using bovine VT oocytes can be successfully performed by co-culturing oocytes with feeder cells that have been preincubated for 15 h and by enucleating and activating oocytes prior to freezing. To improve the survival of frozen–thawed oocytes, we performed co-culture with feeder cells prior to SCNT. We tested the effect of preincubating feeder cells for various amounts of time (2, 5, 15, and 24 h) on the survival of frozen–thawed oocytes and their developmental potential after SCNT. There were differences among the treatment groups (p<0.05). Preincubation of feeder cells for 15 h prior to co-culture improved the survival of frozen–thawed oocytes, but the development potential of these oocytes was not comparable to that of non-V oocytes. Therefore, we enucleated and/or activated oocytes prior to freezing. Following SCNT, the developmental potential of oocytes in the EAVT group was the highest among the vitrification groups and was comparable to that of non-V fresh oocytes. To identify the reason underlying the differences among the VT, EVT, and EAVT groups, we examined ROS production, MAPK activity, and maternal-related gene expression in oocytes and development-related gene expression in blastocysts.
Cryopreservation of oocytes is critical for preserving female genetic resources. Cryopreservation could be a useful technique to provide a steady source of oocytes for SCNT. However, oocytes are more susceptible to cooling damage than embryos. Their large cell size and low permeability to water and CPAs underlie why oocytes are highly sensitive to cryopreservation (Leibo, 1986). In addition, depolymerization of microtubules induced by CPA treatment and cryopreservation causes meiotic spindle disassembly and chromosome misalignment (Shi et al., 2006). Vitrification is a simple, rapid, cost-effective, and reliable method. Many vitrification methods for bovine oocyte cryopreservation have been reported (Dinnyés et al., 2000; Lane et al., 1999; Martino et al., 1996; Vajta et al., 1998). In a previous study (Park et al., 2002), we tested the suitability of the MVC method for SCNT, and showed that by day 8 after SCNT, 8–15% of VT oocytes had developed to the morula stage.
In the present study, we preincubated droplets (10 μL) of feeder cells for various amounts of time and determined how well co-culture with these cells supported the survival of VT oocytes. Co-culture with feeder cells that have been preincubated for 15 h best supported the survival of VT oocytes. Co-culture systems mimic the in vivo environment (Schmaltz-Panneau et al., 2014). Co-culture with somatic cells can overcome the in vitro developmental block of cultured embryos. Co-cultured cells may secrete nutrients, cytotropic factors, or cell-specific glycoproteins (Hwu et al., 1998).
Although blastocysts were produced from VT oocytes following SCNT, freezing of cumulus-enclosed MII oocytes (VT group) was not effective for subsequent SCNT. In the VT group, half of the surviving oocytes were suitable for SCNT; however, enucleation was difficult because the spindle was unclear or undetectable via the Oosight Microscopy Imaging System. In a previous study (Kim et al., 2012), we reported that the Oosight Microscopy Imaging System can be used to observe the meiotic spindles of VT oocytes. Enucleation is critical for SCNT; it must eliminate all genetic material from the recipient cytoplasm without causing parthenogenetic activation (Dominko et al., 2000).
Following co-culture with feeder cells that had been preincubated for 15 h, we compared the SCNT cloning efficiency of various groups of frozen–thawed bovine oocytes, namely, the EVT, EAVT, and VT groups. Significantly more oocytes were suitable for SCNT in the EVT and EAVT groups than in the VT group (p<0.05). Following SCNT, the percentage of oocytes that underwent cleavage in the EAVT group (70.7%) was comparable to that in the non-V group (72.4%) and was significantly higher than that in the VT and EVT groups (p<0.05). In previous studies, following SCNT using VT oocytes, the cleavage rate of reconstructed embryos was reported to be about 50% (Booth et al., 1999; Hou et al., 2005; Yang et al., 2008) and 60–70% (Muenthaisong et al., 2007), and the developmental potential of MII oocytes and enucleated oocytes did not differ (Muenthaisong et al., 2007).
In addition, these studies reported varying percentages of SCNT reconstructed embryos that developed to the blastocyst stage of 7–10% (Booth et al., 1999, used blastomeres; Muenthaisong et al., 2007, used ear fibroblasts; Yang et al., 2008, used fetal fibroblasts) and 16% (Hou et al., 2005, used cumulus cells or ear fibroblasts). In the current study, we used ear fibroblasts as donor cells. The overall cloning efficiency in the EAVT group (22%) was higher than that in the VT (15%) and EVT (14%) groups and was comparable with that in the non-V group. This indicates that SCNT can be performed using enucleated VT oocytes, as well as with non-V fresh oocytes. On the basis of the aforementioned data, we examined the levels of ROS and phosphorylated MAPK as well as gene expression in the various groups.
The ROS level in the EAVT group was similar to that in the non-V group and was significantly lower than that in the VT and EVT groups (p<0.05). Oxidative stress is mediated by ROS, which are by-products of normal cellular metabolism in mitochondria and serve as key signaling molecules in various physiological and pathological processes (Al-Gubory et al., 2010). An increase in ROS production may lead to a decrease in the intracellular adenosine triphosphate (ATP) concentration and the glutathione/glutathione disulfide ratio, as well as a concomitant increase in the cytosolic concentration of calcium ions, which are all detrimental to oocyte health (Tarin, 1996). We expected freezing to affect ROS production in oocytes; however, the level of ROS in the EAVT group was lower than that in the VT and EVT groups, and was similar to that in the non-V group. This result suggests that oocytes were exposed to different levels of oxidative stress among the groups, and that, among the vitrification groups, oocytes in the EAVT group are most suitable for SCNT.
Microfilament localization and the level of phosphorylated MAPK did not vary among the groups and did not change upon freezing. Microfilaments, or actin filaments, are the thinnest filaments of the cytoskeleton, a highly versatile structure that functions in cytokinesis, cell shape changes, cell movement, and cell division (Kim et al., 2000). In the present study, we found no differences in actin filament localization among the groups, except for the absence of chromatin and spindle staining in the EVT and EAVT groups. Protein levels of MAPK and phospho-44/42 MAPK did not vary among the treatment groups. MAPK is a signaling molecule that associates with centrosome components and is related with microtubule stabilization and thereby with the control of spindle assembly and microtubule configuration (Sun et al. 2001).
This is the first report to analyze mRNA expression of maternal and development-specific genes in frozen–thawed SCNT cytoplasts and the resulting SCNT embryos. The mRNA levels of several maternal genes (ZAR1, BMP15, and NLRP5) and the stress gene HSF1 were lower in the VT, EVT, and EAVT groups than in the non-V group (p<0.05). The mRNA levels of the GDF9 (maternal) and SOD1 (antioxidant) genes did not vary between the non-V group and the treatment groups, with the exception that the level of GDF9 mRNA was lower in the EVT group than in the non-V group (Fig. 3C). Accumulated maternal mRNA in the oocyte is crucial for successful embryo development prior to activation of the embryonic genome (Biase et al., 2014).
Among the various genes expressed in germ cells, ZAR1, BMP15, GDF9, and NLRP5 transcripts are detected at much higher levels in oocytes than in gonads and are thought to be oocyte-specific markers (Pennetier et al., 2004). ZAR1 was the first oocyte-specific maternal effect gene to be identified and is critical for early-phase embryo development; knockout of this gene renders embryos incapable of developing beyond the first cleavage (Wu et al., 2003). Similarly, NLRP5 is a maternal effect gene required for early development prior to zygotic genome activation (Tong et al., 2000). The NLRP5 transcript is detected during oocyte growth from primary follicles and accumulates during oocyte development, and its level decreases after fertilization (Hamatani et al., 2006). BMP15 and GDF9, germ cell-specific members of the transforming growth factor-β superfamily, directly affect oocyte growth and function (Dube et al., 1998; McGrath et al., 1995). The present study indicates that oocyte cryopreservation causes the loss of some cytoplasmic mRNA.
Transcription of heat shock genes is rapidly induced after temperature stress. Heat shock gene expression is crucial for the survival of cells exposed to extracellular stress stimuli and also for normal cellular physiology. HSF1-null oocytes have a normal morphology (Pirkkala et al., 2001). SOD1 binds to copper and zinc ions and is one of three superoxide dismutases responsible for destroying free superoxide radicals. The encoded isozyme is a soluble, cytoplasmic/mitochondrial intermembrane space protein that forms a homodimer to convert naturally occurring, but harmful, superoxide radicals into molecular oxygen and hydrogen peroxide (Kostyuk et al., 2004). In the current study, the mRNA levels of HSF1 and SOD1 were lower and higher, respectively, in the vitrification groups than in the non-V group. This showed that VT oocytes survived well when cultured with feeder cells that had preincubated for a long time.
The mRNA expression of developmental potential–related genes (POU5f1, Interferon-tau, and SLC2A5) in SCNT blastocysts was significantly lower in the EVT, EAVT, and VT groups than in the non-V group; the exception was SLC2A5 expression in the EAVT group, which was comparable to that in the non-V group. The higher mRNA expression of POU5f1, Interferon-tau, and SLC2A5 in the EAVT group than in the VT and EVT groups showed that, among the vitrification groups, oocytes in the EAVT group had the highest cloning efficiency. POU5f1 is a master regulator that is expressed at the beginning of mammalian embryogenesis. Variations in the level and pattern of POU5f1 expression might be responsible for at least some of the problems related to cloning (Boiani et al., 2002). Interferon-tau is the primary agent responsible for maternal recognition of pregnancy in cattle (Roberts et al., 1992) and is secreted exclusively by trophectodermal cells of blastocysts. This is consistent with the notion that the mRNA expression of this gene is high in good-quality embryos. The Interferon-tau mRNA level in the EAVT group was higher than that in the EVT and VT groups, although it was still lower than that in the non-V group. SLC2A5 (GLUT5) is a fructose transporter that is expressed in eight-/16-cell-stage embryos (Augustin et al., 2001) and in skeletal muscle, testis, kidney, fat tissue, and brain. The mRNA level of SLC2A5 in the EAVT group was significantly higher than that in the VT and EVT groups, but was not different from that in the non-V group.
The mRNA expression of the proapoptotic gene CASP3 was significantly lower in the EVT and EAVT groups than in the non-V group. Oocytes are exposed to several instances of heat shock stress during the many mechanical and chemical treatment steps involved in SCNT. Heat shock induces apoptosis in preimplantation embryos in a developmentally regulated manner (Paula-Lopes et al., 2002). Hsp70 expression was significantly lower in the EVT and EAVT groups than in the non-V and VT groups, similar to CASP3 expression. These results indicate that enucleated oocytes are safer for the production of SCNT embryos than MII oocytes. Changes in DNA methylation in embryos occur in an extremely organized manner to set up imprinting patterns that are vital for several biological events. Dnmt3A is thought to be responsible for de novo methylation because its activity appears to be targeted to certain domains of the genome (Okano et al., 1999). The Dnmt3A mRNA level was significantly lower in the EVT group than in the non-V group.
This study demonstrates that optimized feeder cell co-culture along with oocyte enucleation and activation prior to freezing can be a novel technique for successful SCNT, resulting in enhanced oocyte survival, low ROS production, and a high cloning efficiency. This finding has important implications for nuclear transfer research and the establishment of oocyte banks for SCNT.
Footnotes
Acknowledgments
This work was supported by grants from the Next-Generation BioGreen 21 Program (PJ009075) and the Cooperative Research Program for Agriculture Science and Technology Development (PJ009103), Rural Development Administration, Republic of Korea.
Author Disclosure Statement
The authors declare that there are no conflicting financial interests.