
Abstract
Abstract
We have characterized the differentiation potentiality and the developmental potential of cloned embryos of fetal bone marrow mesenchymal stem cells (BMSCs) isolated from Mongolian sheep. BMSCs were harvested by centrifuging after the explants method and the mononuclear cells obtained were cultured. The isolated BMSCs were uniform, with a fibroblast-like spindle or stellate appearance, and we confirmed expression of OCT4, SOX2, and NANOG genes at passage 3 (P3) by RT-PCR. We measured the growth of the passage 1, 5, and 10 cultures and found exponential growth with a population doubling time of 29.7±0.05 h. We cultured the P3 BMSCs in vitro under inductive environments and were able to induce them to undergo neurogenesis and form cardiomyocytes and adipocytes. Donor cells at passages 3–4 were used for nuclear transfer (NT). We found the BMSCs could be expanded in vitro and used as nuclear donors for somatic cell nuclear transfer (SCNT). Thus, BMSCs are an attractive cell type for large-animal autologous studies and will be valuable material for somatic cell cloning and future transgenic research.
Introduction
B
Development of somatic cell nuclear transfer (SCNT) technologies has provided a new and faster way to create transgenic animals of many species (Inoue et al., 2003; Lagutina et al., 2007; Ogura et al., 2000; Vajta and Callesen, 2012). The successful transgenic cloning of Mongolian sheep, a species native to the Mongolian plateau, is a major research target because of the strong adaptability of this species. Somatic cloning and transgenic technologies have been widely applied to sheep breeding, and whether donor cell selection can improve the success rate of nuclear transfer to increase transgenic efficiency has become a key research focus (Kassem et al., 2004; Qin et al., 2009).
In this study, we expanded our knowledge of the use of MSCs as donor cells and the application of these cells to somatic cell cloning. Compared with traditional donor cells and fetal fibroblast cells, MSCs with rapid proliferation and antiaging abilities can also shorten nuclear reprogramming time (Blelloch et al., 2006; Bosch et al., 2006; Faast et al., 2006). Thus far, MSCs have been used successfully to clone cows and pigs (Jin et al., 2007; Kato et al., 2004). Using these cells to establish transgenic animals that produce transgenic cell lines is the long-standing goal.
The aims of this study were to establish a culture system of fetal Mongolian sheep BMSCs in vitro, study their multiple differentiation potentiality, and test these BMSCs for use as donor cells and transgenic clones. In doing so, we provide the essential groundwork needed for improving cloning efficiency.
Materials and Methods
All chemicals and culture media used in this study were of cell culture grade and obtained from Sigma Chemicals Co. (St. Louis, MO, USA) unless otherwise indicated. The plasticware was from Nunc (Roskilde, Denmark).
Ethics approval
The study was approved by the animal research ethics board of our university, and return of a survey was considered consent to participate.
Cell isolation and culture
Mongolian sheep 3 months into pregnancy were used. Briefly, after the fetus was removed by cesarean section, the fetal hind legs were cut off and placed onto a clean bench. The leg muscles were removed by sterile gauze, and the femur parts were washed with phosphate-buffered saline (PBS). After cutting the femoral ends, the marrow cavity was flushed by the sterile syringes containing PBS and inserted at one end to collect the bone marrow. The bone marrow was centrifuged at 1000×g for 10 min, the supernatant was removed, and the collected cells were transferred to a clean centrifuge tube and washed two times with serum-free Dulbecco's Modified Eagle Medium (DMEM). The mononuclear cells were collected and cultured with DMEM-F12 supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively) at 37°C in 5% CO2. After 6 h, the medium was changed and nonadherent cells were removed. After 24 h, the medium was changed for the second time. After that, the medium was changed every 2–3 days until the cells had grown to 90% fusion. At that point the cells were digested with 0.25% trypsin and cultured and subcultured 1:3.
Cell doubling method
Passages (P) 1, 5, and 10 of MSCs were seeded into 24-well plates at 1×104 cells/well and grown in 500 μL of the culture medium. The cells in three wells of each group were counted every day for 9 days, and growth curves were drawn. Population doubling time (PDT) was calculated as t[lg2/(lgNt − lgN0)] (N0 and Nt represent the cell counts after inoculation and t the hours after the start of culturing, respectively).
Reverse transcription polymerase chain reaction
Expression of pluripotency was analyzed by reverse transcription polymerase chain reaction (RT-PCR), as described earlier (Yadav et al., 2011). Briefly, total RNA was isolated from Mongolian sheep BMSCs at passage 3 using RNAiso Plus (cat. no. 9108, TaKaRa). The RNA was reverse transcribed into cDNA using the PrimeScript® 1st Strand cDNA Synthesis Kit (cat. no. 6110, TaKaRa). PCR mix (25 μL) was prepared by using cDNA, 10×PCR buffer, 25 mM MgCl2, 10 mM dNTP mix, 10 μM each forward and reverse primers, and Taq DNA polymerase (5 U/μL). The conditions for amplification were 35 cycles consisting of denaturation at 94°C for 30 sec, annealing at X°C (primer specific) for 30 sec, elongation at 72°C for 30 sec, and final extension at 72°C for 5 min. The PCR primers and the reaction conditions are summarized in Table 1. A set of reactions without template cDNA was used as the negative control for PCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used at all stages as a housekeeping marker gene. The amplified DNA fragments were resolved on a 2% agarose gel containing ethidium bromide (final concentration 0.5 μg/mL) and visualized by a gel documentation system (Alpha Imager, Alpha Innotech, San Leandro, CA, USA). The gene-specific bands were excised and purified using an EasyPure Quick Gel Extraction Kit (Beijing TransGen Biotech Co., Ltd.) for further analysis.
Induced differentiation in vitro
P3 cells were cultured in DMEM-F12 until they reached 70–80% confluence in 35-mm dishes. To induce neurogenesis, the undifferentiated stem cells were exposed to neuronal preinduction media consisting of 20% FBS and 1 mM β-mercaptoethanol in DMEM. After 24 h, the cells were washed twice with PBS and placed in 5 mM β-mercaptoethanol (cat. no. 386, Shanghai Chemical) in DMEM without FBS for 6 h (Woodbury et al., 2000). Cell differentiation was assessed morphologically, stained with Toluidine Blue, and observed under a phase-contrast microscope. To induce cardiomyocytes, the cells were treated with 10 μmol/L 5-azacytidine for 24 h.
Next, the medium was replaced with a fresh medium and incubated in a CO2 incubator. The medium was changed once after 3–4 days. At 1 month, the cells were fixed with 0.4% glutaraldehyde for 2 h and washed with PBS. Cell differentiation was assessed morphologically and stained with Periodic Acid Schiff (PAS) (Saftig et al., 2001). To induce adipogenic differentiation, an adipogenic medium [DMEM-F12, 10% FBS, 1 μmol/L dexamethasone, 17 μmol/L pantothenic acid, 5 mmol/L indomethacin, 1 μmol/L insulin, and 0.5 mmol/L 3-isobutyl-1-methylxanthine (IBMX)] was added to the dishes. The presence of intracellular lipid globules indicative of adipogenic differentiaion was assessed by staining the cells with Oil Red O solution on day 9. The medium was replaced twice a week.
Staining method
For Toluidine Blue staining, the cells were rinsed two times with PBS, then fixed in 4% formalin and stained in a Toluidine Blue working solution for 2–3 min. Next, they were washed in running tap water for 2 min, rinsed in distilled water, and observed under the microscope. For PAS staining, the cells were rinsed two times with PBS, fixed in 4% formalin, oxidized in 0.5% periodic acid solution for 5 min, and rinsed in distilled water. They were placed in Schiff reagent for 15 min (the sections become light pink during) and washed in lukewarm tap water for 5 min (the sections turn dark pink immediately). The cells were counterstained in Mayer's Hematoxylin for 1 min, washed in tap water for 5 min, and observed under the microscope. For Oil Red O staining, cells were washed twice with PBS and fixed in 4% paraformaldehyde for 30 min. After fixing, the cells were washed again with PBS and incubated with 0.16% filtered Oil Red O in isopropanol (wt/vol) for 10 min. The Oil Red O stain was aspirated, and the wells were washed with water for 2–3 min before visualizing under a phase-contrast microscope and were imaged immediately after staining.
Specific gene expression detected by RT-PCR
Total RNA was isolated from the treated cells and then reverse-transcribed into cDNA. The induction of specific genes was detected by PCR. ENO2 and GFAP were measured as indicators of neuronal differentiation; NKX2-5 and GATA-4 were measured as indicators of cardiomyocyte differentiation; PPAR and leptin were measured as indicators of adipogenic differentiation. The PCR primers and the reaction conditions are summarized in Table 1.
Nuclear transfer and in vitro culture
Ovaries were collected from slaughtered sheep. Cumulus–oocyte complexes (COCs) were aspirated from follicles 3–8 mm in diameter and transferred to maturation medium. COCs were matured in H-M199 medium, which was supplemented with 10% (vol/vol) FBS, 10 μg/mL follicle-stimulating hormone (FSH), 20 μg/mL luteinizing hormone (LH), 1.5 μg/mL 17β-estradiol (17-βE2), and 0.38 mM sodium pyruvate. Sheep oocytes were cultured at 38.5°C in 5% CO2 in humidified air for 18 h. We selected the oocytes that extruded the first polar body and transferred them to serum-free H-M199 (including 25 mM HEPES, 5 mM sodium bicarbonate), removed the cumulus cells, and then placed them into H-M199 containing 0.25% hyaluronidase for 5 min, followed by cleaning the oocytes.
Denuded metaphase II (MII)-stage oocytes were enucleated by micromanipulation in H-M199 supplemented with 10% FBS, 5 μg/mL cytochalasin B (CCB), and 5 μg/mL Hochest 33342. Briefly, the first polar body and metaphase plate with a small volume of cytoplasm were removed together using a 15-μm beveled micropipette. A single donor cell (which passaged, and reached 95–100% confluence) of approximately 10 μm diameter was used for NT. For fusion, the reconstructed embryos were oriented in a BTX Electro chamber (BTX, Inc., San Diego, CA, USA) filled with 0.3 M mannitol solution containing 0.1 mM MgSO4, 0.05 mM CaCl2, and 0.01% bovine serum albumin (BSA) and pulsed twice with 130 V/mm DC for 30 μsec using a BTX Electro Square Porator (ECM 830, BTX, Inc., San Diego, CA, USA).
After fusion, reconstructed embryos were cultured in 50-μL drops of H-M199 medium supplemented with 7.5 μg /mL CCB at 38.5°C in a humidified atmosphere of 5% CO2 in air for 4 h. For activation, reconstructed embryos were treated with 5 μM ionomycin in H-M199 medium for 5 min, followed by 4 h of culture in 2 mM 6-dimethylaminopurine (6-DMAP) in medium synthetic oviductal fluid (SOF), supplemented with minimum essential medium (MEM), essential and nonessential amino acids, and 4 mg/mL BSA (m-SOFaa). Reconstructed embryos were cultured in a humidified atmosphere of 5% CO2 in air for 5–8 days.
Statistical analysis
Data analysis was performed using SPSS 9.2. The effects of different cryopreservation media on prefreezing and postthaw viability of cells were tested by one-way analysis of variance (ANOVA).
Results
Isolation and culture of Mongolian sheep BMSCs
After isolation, the cells were plated into two 25-cm2 flasks. After 6 h, the cells, which were irregular in shape, began to attach to the plastic. Most of the nonadherent cells were removed during the second medium change at 24 h, and primary cultures could be initiated successfully. The number of cells increased and the majority were composed of cells with a characteristic spindle shape 48 h later (Fig. 1A). After the medium was changed, the cells became confluent. The number of colonies increased gradually to 80% confluence after 6–7 days of seeding (Fig. 1B). After two to three subcultures, cell morphology was consistent and had a uniformly long fusiform shape; 4–5 days later, cells reached 80% confluence (Fig. 1C). The cell lines at the third passage were frozen in liquid nitrogen for longer storage and future use.
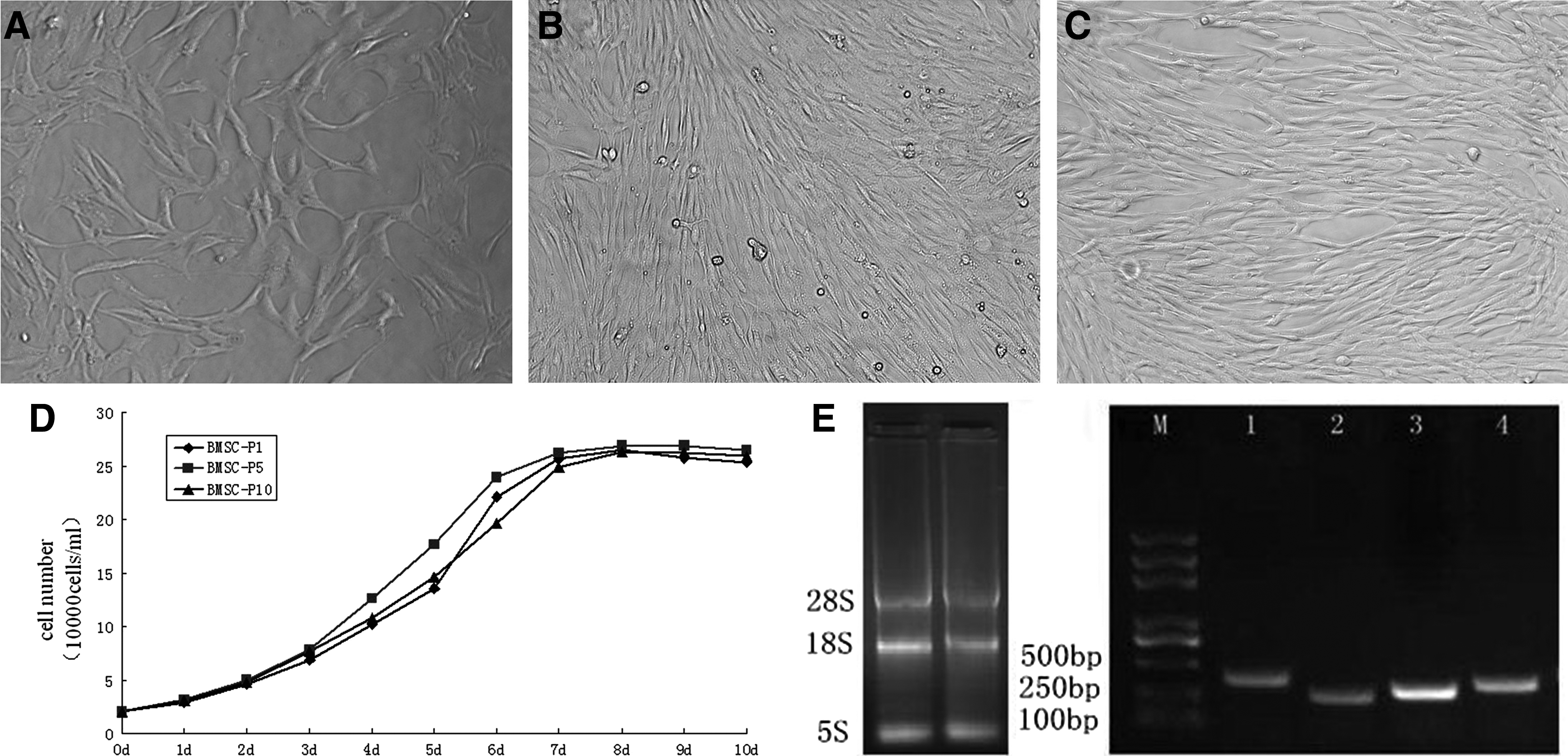
Fetal Mongolian sheep BMSCs in culture. (
Characterization of Mongolian sheep BMSCs
Cell growth curves were generated for Mongolian sheep fetal BMSCs at P1, P5, and P10, separately. The results demonstrated that P1, P5, and P10 growth curves shared a similar S-shape (Fig. 1D). They all showed an incubation period of 1–2 days and slow growth. At the logarithmic growth phase after day 3, the cells entered a rapid growth phase until leveling off at days 7–8, when the cell proliferation rate slowed down. The cells of P5 grew strongly compared with those of P1 and P10. The average population doubling time of the Mongolian sheep fetal BMSCs was calculated to be 29.7±0.05 h. We extracted total RNA of P3 BMSCs and detected three bright bands by electrophoresis corresponding to 28S, 18S, and 5S. We then used RT-PCR and measured expression of OCT4, SOX2, and NANOG to characterize the stemness of BMSCs (Fig. 1E). The resulting sequences were aligned and analyzed using the online Basic Local Alignment Search Tool (BLAST) as described by Yadav et al. (2011). All sequences showed 100% homology with Ovis aries homeobox transcription factor mRNA.
Neural differentiation of BMSCs
After treatment with noninducing medium, no obvious changes were observed in BMSC morphology. After the BMSCs were treated with neural differentiation-induced medium for 3 h, we found that the cells improved their refractivity and began to shrink and become more rounded. At 12 h later, some neurites appeared on cell somas, with branched ends and inflated tips. Some ends contacted other cell somas or neuritis and neural synaptic structures could be observed (Fig. 2A). After 24 h, the cells became bipolar, multipolar, conically shaped, and differentiated into neuron-like cell morphology. Most of the cells were intertwined. After these differentiated cells were stained with Toluidine Blue, the Nissl bodies were visible as deep blue particles or plaque, and the nuclei appeared blue (Fig. 2B). No Nissl bodies were observed in the control group after staining (Fig. 2C). We found by RT-PCR that ENO2 and GFAP, genes specific for neural differentiation, were expressed (Fig. 3A). Together, these results indicate the BMSCs are able to differentiate into neural lineage cells.
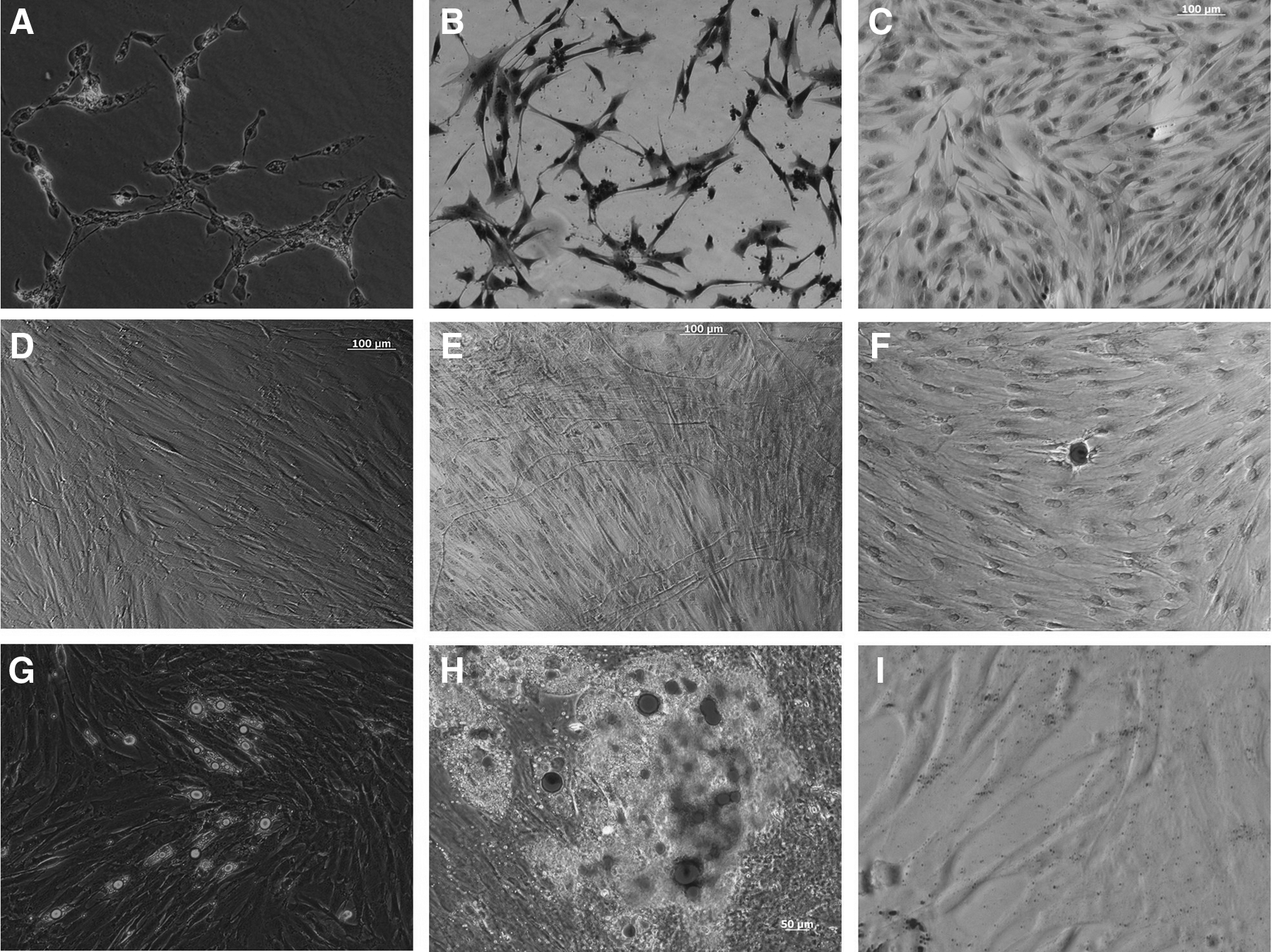
Staining identification of neural (

Gene expression profile of differentiation research. (
Cardiomyocytes differentiation of BMSCs
After the BMSCs were induced by 5-azacytidine (5-aza) for 24 h, the arrangement and the morphology of cells began to change. After 1 week of induction, cell volume increased, and multispindles were arranged in parallel (Fig. 2D). After 2 weeks, the cells became short columns and protruded at both ends; the adjacent cells processed in close contact, and PAS staining showed a large amount of glycogen accumulation. After 3 weeks, the short columnar cells were interconnected, whereas some cells were myotube-like. After 4 weeks, the cells were smaller and in a multinucleated myotube-like structure. PAS staining showed glycogen accumulation around the myotube-like structure (Fig. 2E), but there no spontaneous pulsation phenomenon observed under the phase-contrast microscope. PAS staining showed glycogen accumulation in the control group. In separate experiments, we used RT-PCR to confirm NKX2-5 and GATA-4 expression for cardiomyocytes after 28 days of additional culture(Fig. 3B). These observations confirm that BMSCs can be chemically transformed into cardiomyocytes.
Adipogenic differentiation of BMSCs
When cultured under adipogenic conditions, the cells differentiated into adipocytes and exhibited a high intensity of Oil Red O stain in the cytoplasm of cells 9 days postinduction, signifying the presence of lipid globules (Fig. 2G–H). RT-PCR showed expression of PPAR and leptin, genes specific for adipogenic differentiation (Fig. 3C).
Nuclear transfer experiments
BMSCs were used as nuclear donors to produce cloned sheep embryos with a cleavage rate of 79.8% (138/173). The efficiency of development to the blastocyst stage was 1.73% (3/173) (Fig. 4A–C). This shows that the Mongolian sheep fetal MSCs can be used as nuclear donors and develop to the blastocyst stage. We also used sheep fibroblast cells (SFCs) as donors to perform the nuclear transfer (NT) experiment. The cleavage rate was 75.7% (187/247) and the blastocyst rate was 1.2% (3/247) (Fig. 4D–F).
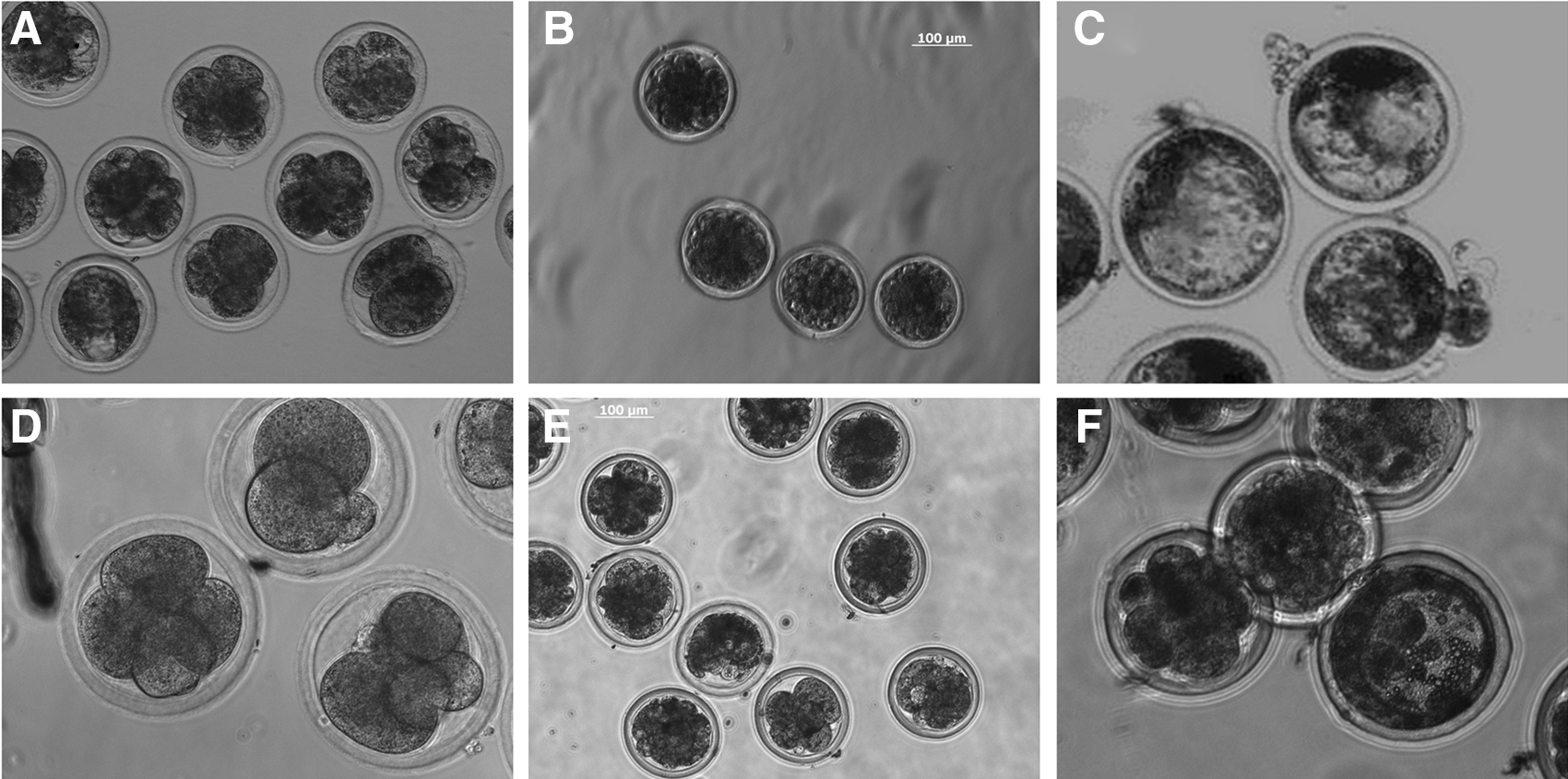
SCNT (
Discussion
The fetal MSC lines isolated in this study were grown using culture and differentiation protocols reported previously (Bobis et al., 2006; Kassem et al., 2004). In recent years, MSCs have become important seed cells for tissue engineering because of their abundant source and convenience. Most of this work has relied on adult BMSCs, with few studies using fetal BMSCs. Here we successfully isolated and characterized the differentiation potential of fetal bone marrow MSCs, and in doing so, laid the foundation for future clinical applications.
The most common method for MSC isolation is based on the ability of MSCs to adhere to plastic surfaces selectively, and the commonly used method is gradient centrifugation. In this study, we instead isolated fetal BMSCs by a whole bone marrow adherent method. For the isolated cell morphology, uniform surface marker in detecting molecules, less than 95% of the cells expressed CD34 hematopoietic stem cell lineage, and more than 98% of the cells showed no expression of lymphocytic lineages of CD45 (Fukuchi et al., 2004; Soleimani and Nadri, 2009). After 6 h, the cells began to attach to the plastic; after the second medium change at 24 h, the BMSCs were highly pure and exhibited strong proliferation and differentiation abilities after repeated passage in vitro. After increased culture time and repeated passaging, the cell morphology was a consistent and uniformly long fusiform shape, similar to the description by Vacanti et al. (2005), Engler et al. (2006), and Branch et al. (2012). The growth curves of MSCs are similar with an S-shape. During the exponential phase, the average PDT for P1–P10 MSCs was 29.7±0.05 h.
Transcription factors that play critical roles in maintaining pluripotency of stem cells have been identified, including OCT-4, NANOG, SOX2, STAT 3, c-Myc, and KLF 4. Expression of these genes is used to characterize stem cells in various species (Anja and Rolf, 2013; Niwa et al., 2007). Here, we measured expression of OCT4, SOX2, and NANOG to characterize the stemness of P3 BMSCs. RT-PCR showed that these genes are expressed positively, in agreement with Violini et al. (2009) who detected expressions of these genes in porcine bone marrow (BM)-derived cells. Expression of these genes was also reported in human amniotic epithelial-derived cells, equine umbilical cord and umbilical cord matrix cells, buffalo amniotic fluid, and fetal skin-derived fibroblast cells (Cremonesi et al., 2008; Carlin et al., 2006; Miki et al., 2005; Yadav et al., 2011, 2012a). The phenotypes of MSCs are usually characterized by using immunocytochemical detection or fluorescence-activated cell sorting (FACS) of cell-surface molecule expression. However, the lack of specific markers renders this characterization difficult to interpret.
MSC stem cells are derived from the mesoderm, but their differentiation ability is not limited to the mesoderm. In this study, we successfully performed cross-mesoderm–inducing differentiation for BMSCs. For inducing differentiation of the ectoderm neural cells, BME is capable of supporting the viability and differentiation of fetal mouse brain neurons (Ishii et al., 1993) and was used as an effective inducer of neural differentiation in MSCs (Suzuki et al., 2004; Woodbury et al., 2000).
Nestin expression is a necessary stage of neural differentiation of MSCs, and serum in culture medium can inhibit nestin expression (Wislet et al., 2003, 2005). We found that BMSCs could differentiate into neural cells under serum-free conditions, and expression of ENO2 and GFAP could be detected by RT-PCR, as found by Woodbury (Woodbury et al., 2000). Thus, the BMSCs successfully underwent ectoderm differentiation.
5- aza is commonly and efficiently used as a cardiomyocyte differentiation induction medium. An analog of cytidine that leads to DNA demethylation, 5-aza causes an initial rapid increase of growth (Gaustad et al., 2004; Xu et al., 2004). Following this increase, however, cells are maintained for a long time without detachment from the surface of the culture dish (Zhang et al., 2005). In this study, PAS staining showed a large amount of glycogen accumulation, followed by interconnection of cells and formation of a multinucleated myotube-like structure. These results were similar to those of Rangappa et al. (2003). Zhang (Zhang et al., 2005) found a spontaneous pulsation phenomenon in induced cells, but we did not observe this in our experiments. Morphological observation of specific gene expression by RT-PCR indicates that 5-aza promoted cardiomyocyte differentiation.
Indomethacin is the rapidest inducer of adipogenic differentiation. We find that after 3–4 days of treatment, small oil droplets were observed under the inverted microscope. After 7 days of incubation, Oil Red O staining revealed red oil vacuoles in the cytoplasm; after 12 days, we saw formation of large lipid droplets, as previously reported in adipogenic-induced human MSCs (Dominici et al., 2006; Franco et al., 2009). The expression of the adipogenic differentiation-specific genes PPAR and Leptin was confirmed by RT-PCR, showing that BMSCs can differentiate to adipocytes.
Finally, in our NT experiments, we demonstrated fetal Mongolian sheep BMSCs can give rise to cloned blastocysts at a higher development rate. Although preimplantation development is not necessarily a reliable predictor of successful postimplantation development, this higher development rate may be due to the stem cell nature of the cell donor nucleus (Colleoni et al., 2003; Peterbauer-Scherb et al., 2010). In our experiments, the blastocyst rate is generally low, but this may be due to experimental error with this challenging technique. However, we did obtain cloned blastocysts, suggesting that fetal Mongolian sheep BMSCs can be expanded in vitro and used as nuclear donors to produce SCNT embryos.
Conclusions
We found that fetal Mongolian sheep BMSCs can be cultured in vitro, exhibit good growth, and have a high proliferation ability. The expression of pluripotency marker genes showed that these cells have the characteristics of stem cells and could be induced to differentiate into neurogenesis, cardiomyocytes, and adipocytes in vitro, suggesting that these cells have the capacity to differentiate into cells of different germ layer lineages. The BMSCs can be expanded in vitro and used as nuclear donors to produce SCNT embryos. Thus, BMSCs are an attractive cell type for large animal autologous studies and will be valuable for somatic cell cloning and transgenic research.
Footnotes
Acknowledgments
The authors would like to thank Professor Huanmin Zhou for providing the Mongolia sheep and Dr. Yiyi Liu and Lu Li for their technical help. This study was supported by a Grant-in-Aid for the major project of Inner Mongolia Natural Science Foundation (A) (2012ZD03), a Grant-in-Aid for China Agricultural University cooperation projects (B) (2010), and the Inner Mongolia Key Laboratory of Biomanufacture.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.