
Abstract
Abstract
Human alveolar type II (AT II) epithelial cells are valuable for the cellular therapy of lung disease. Human induced pluripotent stem cells (iPSCs) have the ability to generate AT II cells that can be used in modeling and treatment of lung disease caused by dysfunction of AT II cells. In this study, we present a simple, effective, and noninvasive way of obtaining human iPSCs from exfoliated renal epithelial cells, which exist in urine. Alkaline phosphatase (AP) staining, immunofluorescence staining, karyotyping, and teratoma experiments have proved that these iPSCs are pluripotent. Urinary iPSCs (UiPSCs) can differentiate into AT II cells with our four-step induction protocol. These cells have phenotypic properties similar to mature human AT II cells, such as outstretched and epithelium-like morphology and the specific expression markers of AT II cells (surfactant proteins A, B, and C). This study indicates that AT II cells can be generated from UiPSCs and these cells may be useful for the study of human lung development and regenerative medicine.
Introduction
H
Induced pluripotent stem cells (iPSCs) are a type of novel stem cell that can be generated from adult somatic cells through reprogramming by introduction of specific genes encoding transcription factors (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). These autologous cells proliferate and differentiate just like embryonic stem cells (ESCs) and could be used for transplantation without the risk of immune rejection. Human iPSCs can be reprogrammed from multiple cell types, including peripheral blood, skin fibroblasts, hepatocytes, etc. Compared with these cells, urinary cells are collected simply in a cost-effective and noninvasive manner. They can be obtained at any age and from any gender and race, except in cases of renal failure (Xue et al., 2013; Zhou et al., 2011). The generation of mature AT II epithelial cells derived from differentiated murine and human iPSCs has been reported in several studies (Ghaedi et al., 2013; Huang et al., 2014; Wong et al., 2012; Zhou et al., 2014). However, there is little information about human urine-derived iPSCs differentiating to AT II epithelial cells. In our study, a four-stage differentiation protocol was used to study the possibility of generation of AT II cells from differentiation of human UiPSCs.
Materials and Methods
Urine collection and UiPSCs generation
All six donors in our study signed a consent form that was available as a requirement for human study. Donors washed their urethral area, and the first micturition of the day was avoided routinely. Only the middle stream of the urine was collected into sterile containers (500 mL; Corning, NY, USA). The volume of samples was at least 200 mL. Urine specimens were later transferred into 50-mL tubes (Corning), and these samples were centrifuged at 300 × g for 10 min at room temperature. The supernatant was carefully discarded, leaving approximately 5 mL of urine in the tube. The cells were resuspended individually, and all of the 50-mL tubes derived from one sample collection were pooled into one single 50-mL tube. The samples were centrifuged at 300 × g for 10 min. The supernatant was discarded and 3 mL of Urineasy Medium (Cellapy, Beijing, China) was added to resuspend the cell pellet. The cells were then transferred onto Matrigel-coated (BD Biosciences) 3.5-cm culture plates (Corning) in 1 mL of Urineasy Medium. The cells were incubated, and the medium was changed every other day.
Urine cells at passage 2 were infected with episomal plasmids containing the human transcription factors Klf4, Oct4, c-Myc, and Sox2 (Invitrogen). Visible cell colonies appeared after 5 days. On day 16, these colonies, which were called urinary iPSCs (UiPSCs), were picked mechanically and expanded in human PSCeasy Medium (Cellapy, Beijing, China) on Matrigel. Reprogramming efficiency was determined by alkaline phosphatase (AP) staining; positive clones with human ESCmorphology comprised about 0.15%. Immunofluorescence staining, karyotyping, and teratoma experiments were done to identify UiPSCs characteristics (Zhou et al., 2011).
Differentiation of UiPSCs
Our differentiation protocol, which generated UiPSCs, included four stages as previously described with slight modification (D'Amour et al., 2005; Huang et al., 2014).
Stage 1
This stage included 24 h primitive streak formation and 3 days of definitive endoderm induction. UiPSCs were treated with EDTA (3 min at 37°C) into three to 10 small cell clumps and plated onto a 10-cm dish (Corning) for 24 h to form embryoid bodies (EBs) in Serum-Free Differentiation (SFD) Medium. The components of SFD Medium are Dulbecco's Modified Eagle Medium (DMEM)/F12 (1:1) (HyClone) supplemented with B27 (Gibco), N2 (Gibco), Glutamax (2 mM, Life Technologies), 1% penicillin-streptomycin (HyClone), 0.05% bovine serum albumin (BSA) (Life Technologies), ascorbic acid (50 μg/mL; Sigma), and monothioglycerol (0.4 μM; Sigma). EBs were then collected and resuspended in primitive streak formation medium containing SFD Medium with addition of Y-27632 (10 μM; Tocris) and human bone morphogenetic protein 4 (BMP4, 3 ng/mL; Tocris) for 24 h. EBs were replated in definitive Endoderm Induction Medium with Y-27632 (10 μM; Tocris), human BMP4 (0.5 ng/mL; R&D Systems), human basic fibroblast growth factor (bFGF, 2.5 ng/mL; Tocris), human activin A (100 ng/mL; Tocris), and SFD Medium for 3 days on a 10-cm dish (Corning).
Stage 2
This stage is 2 days of anterior foregut endoderm induction. On day 5, EBs were replated onto Matrigel-coated 6-cm culture plates in SFD Medium supplemented with dorsomorphin dihydrochloride (1.5 μM; Tocris) and SB431542 (10 μM; Sigma) for 24 h and then changed to 24 h of IWP2 (1 μM; Tocris) and SB431542 (10 μM, Sigma) treatment.
Stage 3
This stage included lung progenitor induction during the next 7–15 days. The resulting anterior foregut endoderm was treated with SFD Medium containing human fibroblast growth factor-7 (FGF-7, 10 ng/mL; R&D Systems), human FGF-10 (10 ng/mL; R&D Systems), human BMP4 (10 ng/mL; R&D Systems), CHIR99021 (3 μM, LC Laboratories), all-trans retinoic acid (ATRA) (1 μM; Sigma), and murine epidermal growth factor (EGF, 20 ng/mL; PeproTech, Inc.) for 8 days.
Stage 4
Stage 4 was days 15–25 of AT II epithelial maturation induction. On day 15, the lung progenitor cells were incubated in EDTA at 37°C for 3 min. EDTA was then removed by suction and SFD Medium was added into the 6-cm culture plate. Cells were separated into cell clumps with a 1-mL pipette tip, replated into Matrigel-coated 6-cm culture plates in SFD Medium supplemented with human FGF-7 (10 ng/mL; R&D Systems), human FGF-10 (10 ng/mL; R&D Systems), and CHIR99021 (3 μM, LC Laboratories), and cultured for for 10 days.
For the control group (UiPSC-con), normal human UiPSC culture medium was used throughout the four steps as a time control compared to the differentiation groups. The cells of differentiated day 15 (Diff. 15), 25 (Diff. 25), and the control group were collected and preserved for the following analysis of real-time PCR and immunofluorescence staining.
Immunofluorescence staining
The cells cultured in four-well plates were first fixed in 4% paraformaldehyde (PFA; Sigma) for 20 min at room temperature and then washed once with phosphate-buffered saline solution (PBS; HyClone). The cells were then permeabilized with 0.5% Triton for 15 min and blocked in 3% BSA (Sigma) for 1 h at room temperature. The cells were stained with the following primary antibodies: NANOG (rabbit, 1:100; Abcam), OCT4 (mouse, 1:100; Abcam), SSEA-4 (mouse, 1:100; Abcam), TRA-1-60 (mouse, 1:100; Abcam), TRA-1-81 (mouse, 1:100; Abcam), SOX2 (rabbit, 1:1000; Abcam), FOXA2 (goat, 1:40; R&D Systems), SOX17 (mouse, 1:50; Abcam), NKX2.1 (rabbit, 1:100; Abcam), PAX9 (rat, 1:200; Abcam), TBX1 (rabbit, 1:100; Abcam), pro-SPB (rabbit, 1:300; Millipore), pro-SPC (rabbit, 1:300; Millipore), SPA (rabbit, 1:100; Santa Cruz Biotechnology, Inc.), SPB (rabbit, 1:100; Seven Hills Bioreagents), and SPC (rabbit, 1:50; Santa Cruz). The secondary antibodies used were goat anti-mouse immunoglobulin G (IgG)-Alexa Fluor 488, goat anti-rabbit IgG-Alexa Fluor 488, goat anti-rat IgG-fluroescein isothiocyanate (FITC) and rabbit anti-goat IgG-FITC, all from Zhongshan Golden Bridge Biotechnology (China).
After blocking, all the single stainings were performed by incubating primary antibodies according to the dilution factors in the staining solution (3% BSA) at 4°C overnight (minimum 12 h), followed by washing three times for 5 min with PBS. The cells were then incubated with the related secondary antibodies at 1:100 dilutions in staining solution in the dark at room temperature for 1 h, washed three times for 5 min, and incubated with Hoechst 33342 (Beyotime, China) solution (diluted as 1:1000 in PBS) in the dark at room temperature for 20 min. The Hoechst solution was then removed by suction, and Antifade Mounting Medium (Beyotime, China) was added to the samples after washing once in PBS. Samples were imaged using an Olympus IX71 microscope coupled with a fluorescent module.
Quantitative real-time PCR
TRIzol, chloroform, and isopropanol in the proportion of 5:1:2.5 was used to isolate total RNA and SNOVart Reverse Transcriptase (SNOVA) was used for reverse transcription. The PCR reaction system contained primers (Table 1), template, double-distilled H2O, and SYBR Green (TIANGEN, China). The conditions were 95°C for 15 min followed by 40 cycles of 95°C for 10 sec and 61°C for 40 sec. Melting curves were obtained for each of the genes. Semi-real-time PCR was performed on an Eco™ Real-Time PCR system (Illumina). Specific markers of human AT II epithelium cells (SPA, SPB, and SPC) were analyzed. Average cycle threshold (CT) values of tested genes were calculated against average glyceraldehyde 3-phosphate dehydrogenase (GAPDH) CT values from the same sample. ΔCT = CT (tested gene) − CT (GAPDH) and ΔΔCT = ΔCT (differentiation group) − ΔCT (control group). The fold changes of gene transcript levels between differentiation group and control group were calculated as 2−ΔΔCT. Statistical analysis was done by one-way analysis of variance (ANOVA) with Bonferroni post hoc comparisons.
Results
Morphology of urinary cells and characterization of UiPSCs
The fresh urine samples after the collections consisted of squamous cells. These cells grew with adherence on days 3–5 and were expanded on days 5–7. The cells proliferated steadily at passage 1 (Fig. 1). The urine cells at passage 2 were transfected with episomal plasmids containing the cDNAs of human Klf4, Oct4, c-Myc, and Sox2. Human ESC-like colonies first appeared on days 5–10 after infection. From day 15 to day 20, those colonies that were large enough and typical as human ESCs-like (flat morphology with defined borders and large nuclei containing prominent nucleoli) could be picked and then expanded steadily at passage 3 (Fig. 2).
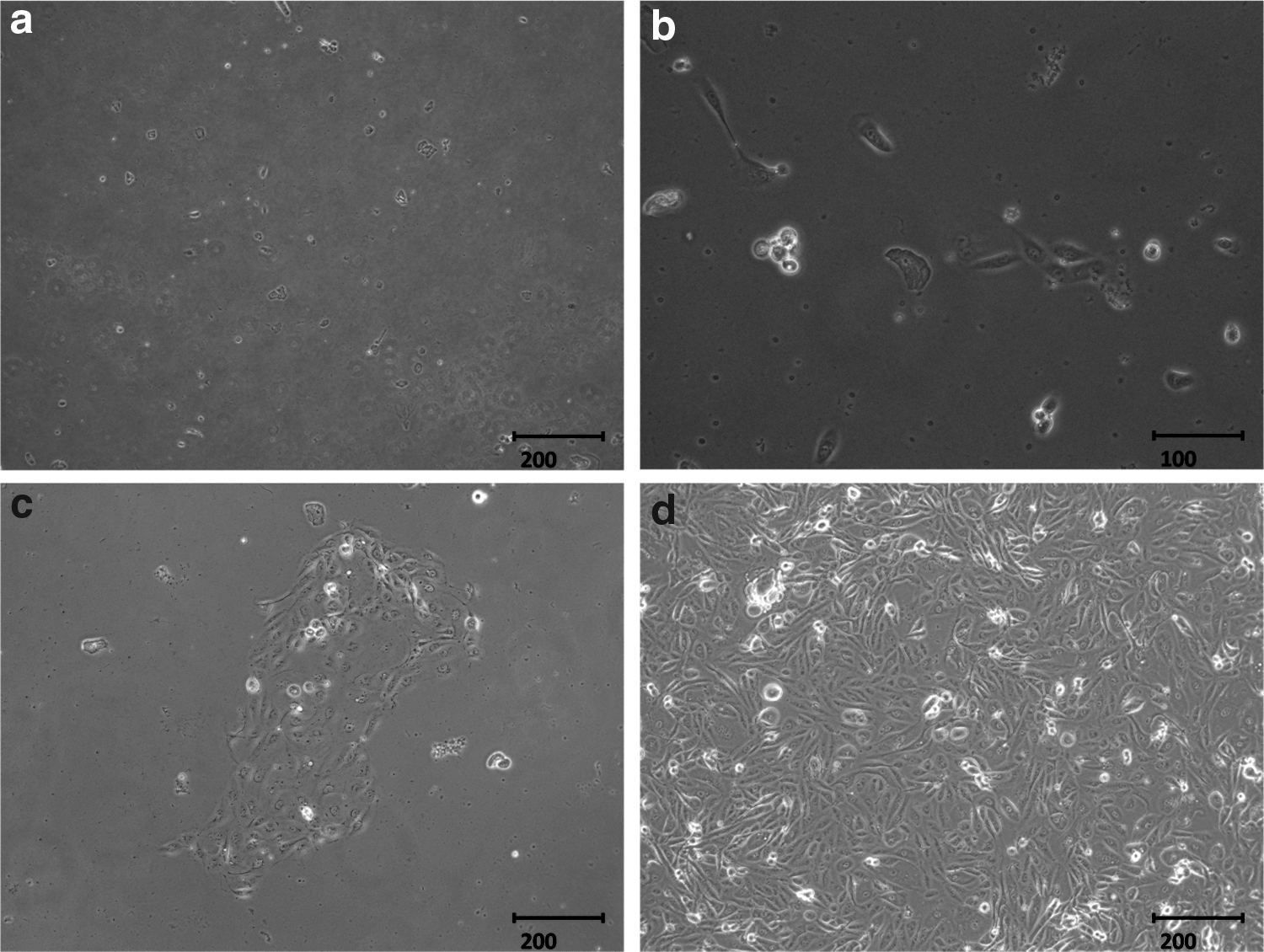
Morphology of urinary cells at different time points after collection. (
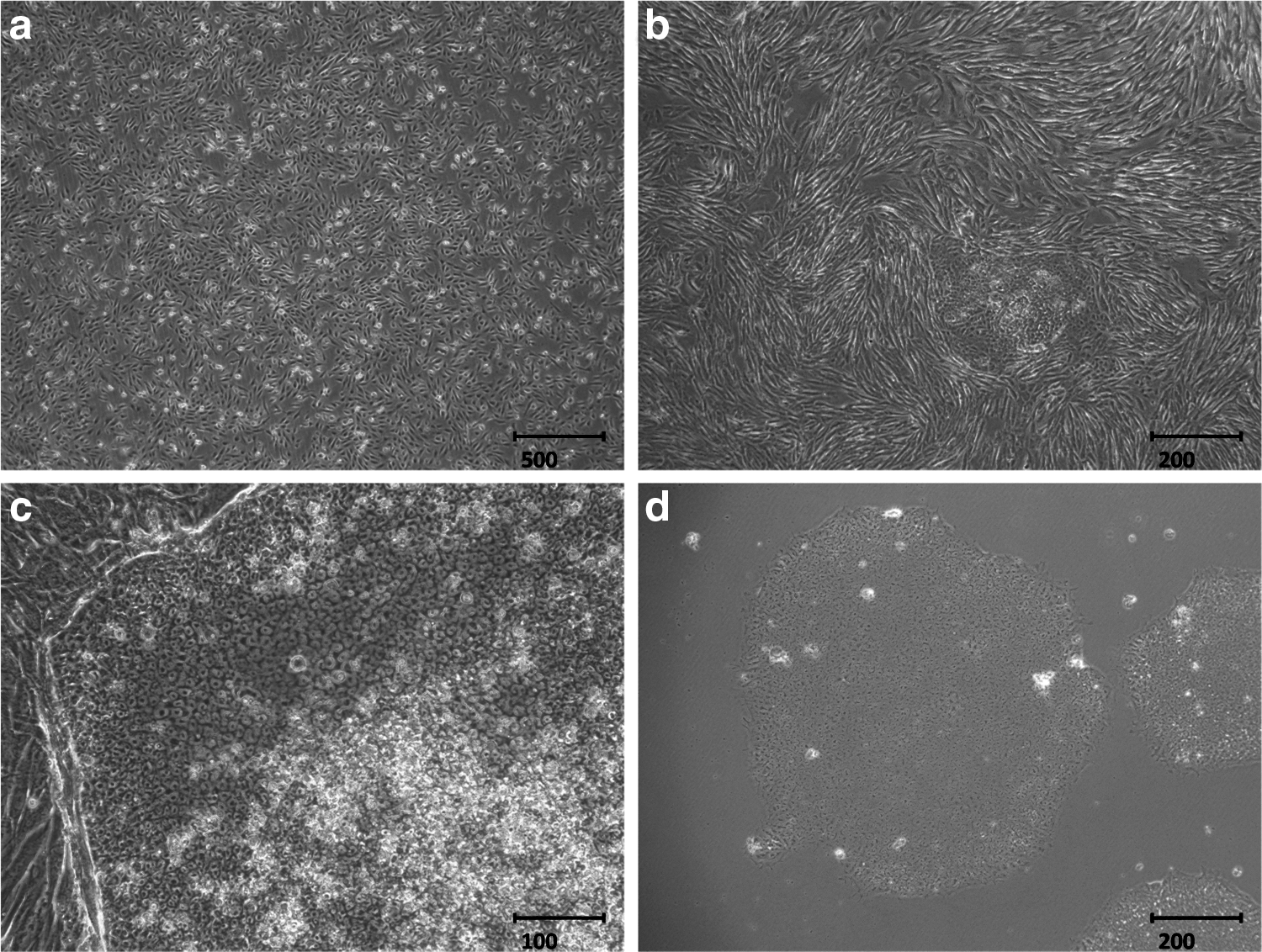
Plasmid infection and UiPSCs generation. (
We analyzed the UiPSCs for their pluripotent properties, both in vitro and in vivo. The AP activity, immunofluorescence staining of pluripotency markers, and karyotyping were in vitro assays. The UiPSCs showed high levels of AP activity, and immunofluorescence staining confirmed expression of pluripotency markers in these UiPSCs, including OCT4, SOX2, NANOG, SSEA4, TRA-1-60, and TRA-1-81 (Fig. 3). Karyotypes of the UiPSCs were normal (Fig. 4). Spontaneous teratoma formation was the in vivo assay for testing the differentiation capability of the UiPSC line (Fig. 5) (Huangfu et al., 2008).
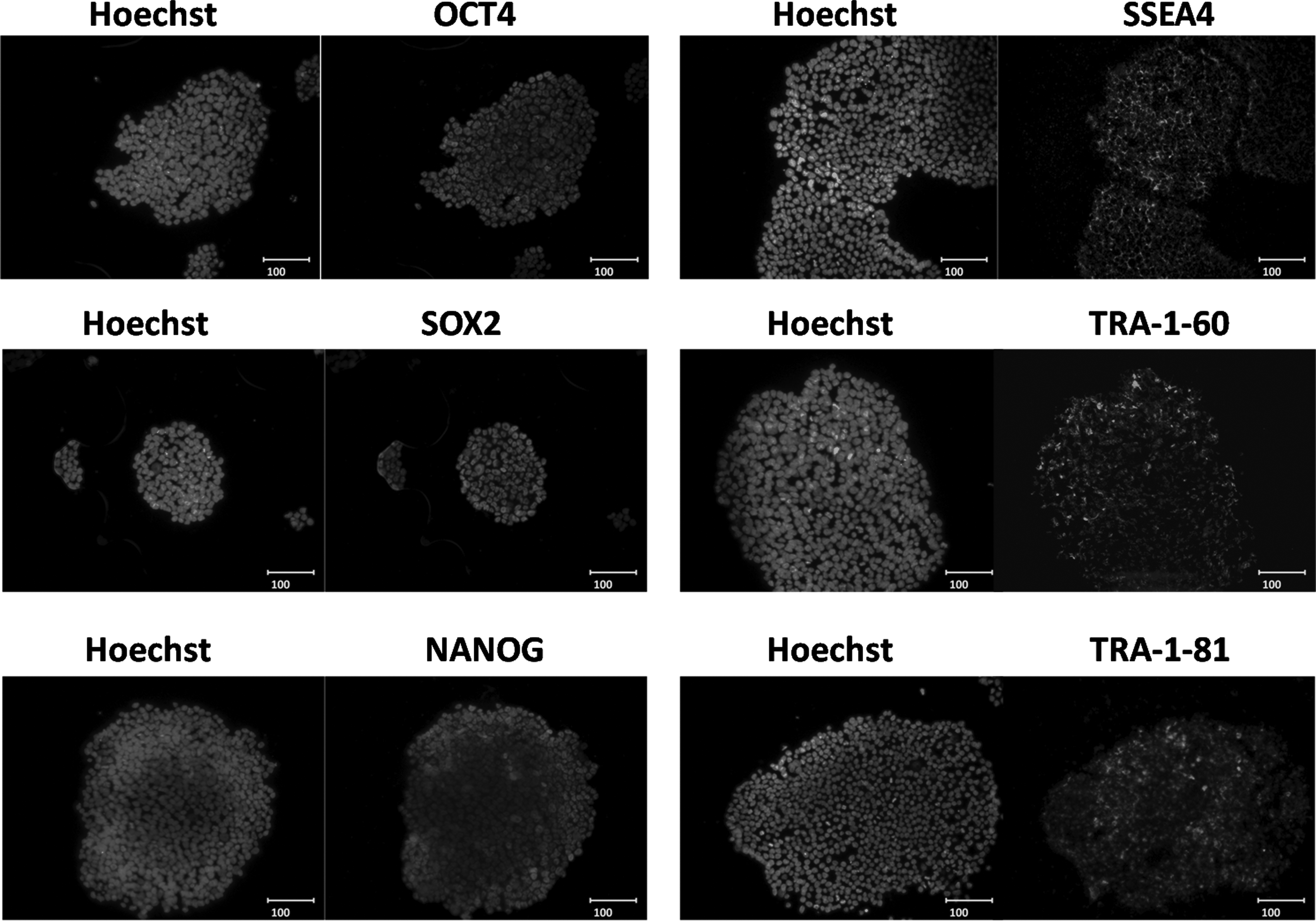
Immunofluorescence staining of UiPSCs using antibodies against OCT4, SOX2, NANOG, SSEA4, TRA-1-60, and TRA-1-81. Cell nuclei were counterstained with Hoechst. All scale bars, 100 μm.
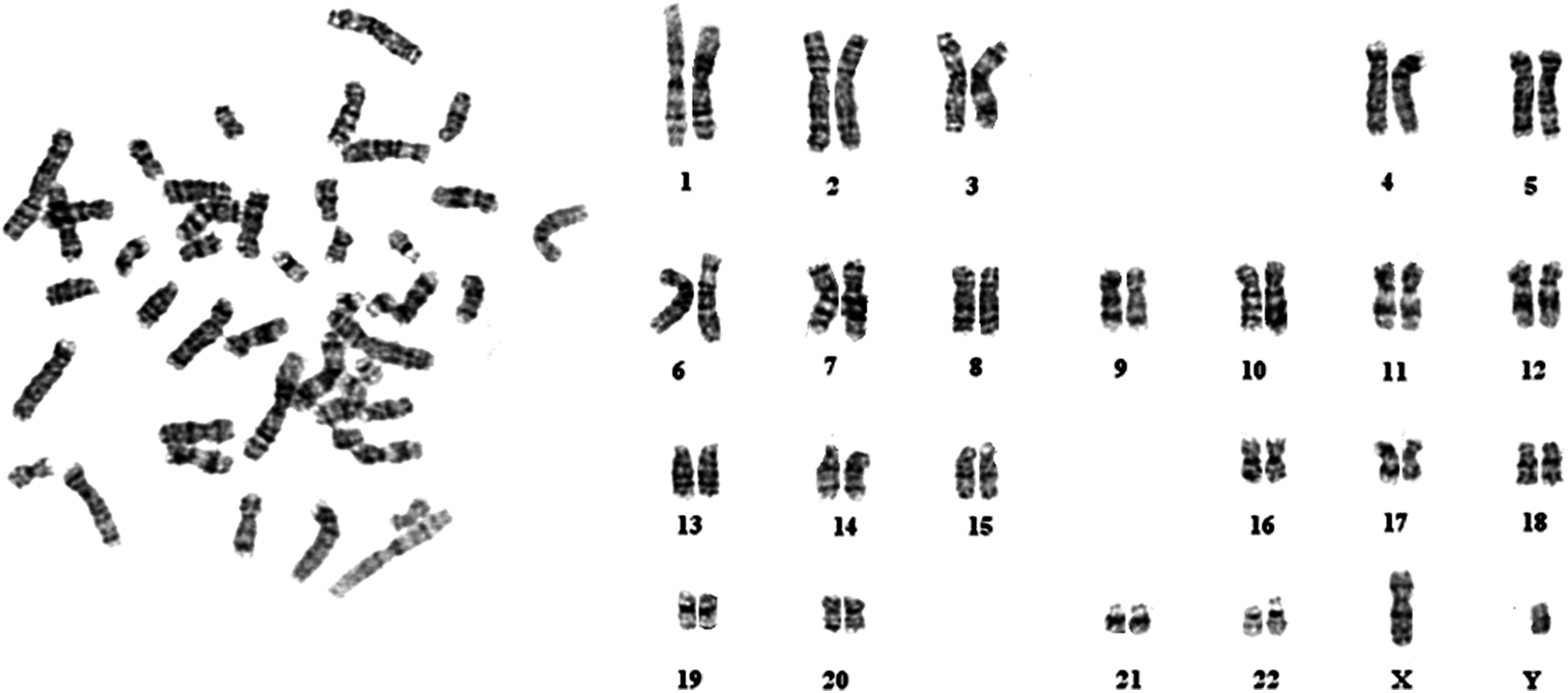
Karyotype analysis of UiPSCs (passage 10) showing a normal karyotype of 24 chromosomes.

Histology of teratoma tissue produced by injecting UiPSCs (passage 17) into severe combined immune deficiency mice. Sections were stained with Hematoxylin & Eosin.
Cellular morphology and immunofluorescence staining during the procedure of differentiation
The differentiation protocol comprised four stages. In stage 1, EBs formed gradually in SFD Medium after UiPSCs were treated with EDTA. The cells grew radially after the adherence of EBs, proving the differentiation of EBs. Then the cell density kept increasing and the cells expressed the specific markers of definitive endoderm cells (FOXA2 and SOX17) (Fig. 6a). In stage 2, immunofluoescence staining for the specific expression markers of anterior foregut endoderm cells (NKX2.1, PAX9, and TBX1) was conducted (Fig. 6b). The anterior foregut endoderm cells further differentiated in stage 3, and these cells might be the lung progenitor cells that expressed marks of pro-SPB and pro-SPC (Fig. 6c). The outstretched and epithelium-like morphology of differentiated cells appeared during stage 4. They could possibly be AT II epithelium cells that showed positive staining for SPA, SPB, and SPC. The percentage of SPA-, SPB-, and SPC-positive cells was about 70% (Fig. 6d). It was determined by visually counting 1000 cells on the basis of Hoechst staining in the differentiated cultures on day 25 (Yan et al., 2014).
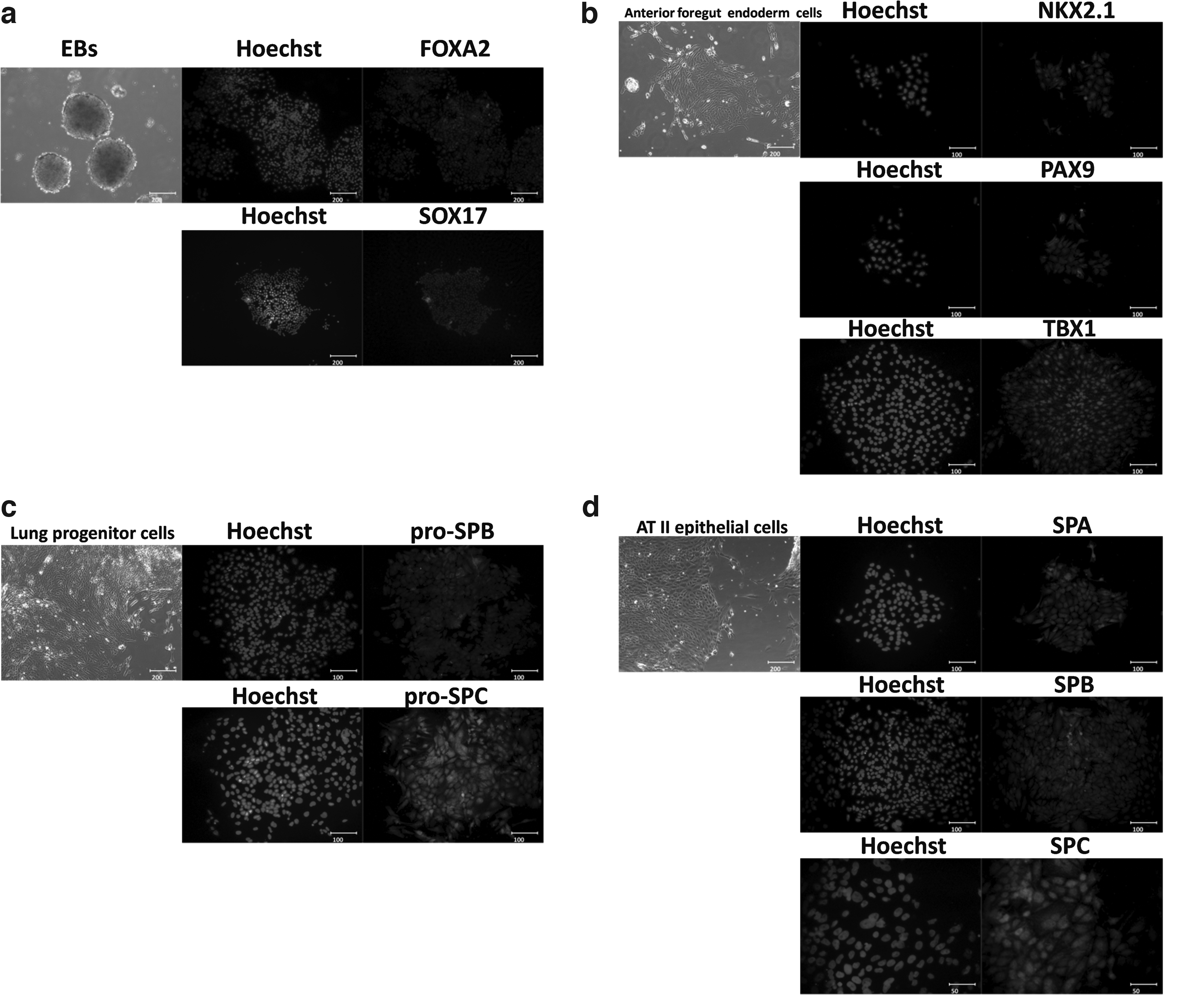
(
Quantitative RT-PCR
Quantitative RT-PCR analysis in differentiated day 15 (Diff. 15 group) and 25 (Diff. 25 group) cells compared with undifferentiated UiPSCs (UiPSCs-con group) were performed from three independent experiments. After differentiation, the specific gene expression of human AT II epithelial cells (SPA, SPB, and SPC) increased significantly in stage 4 (Fig. 7).
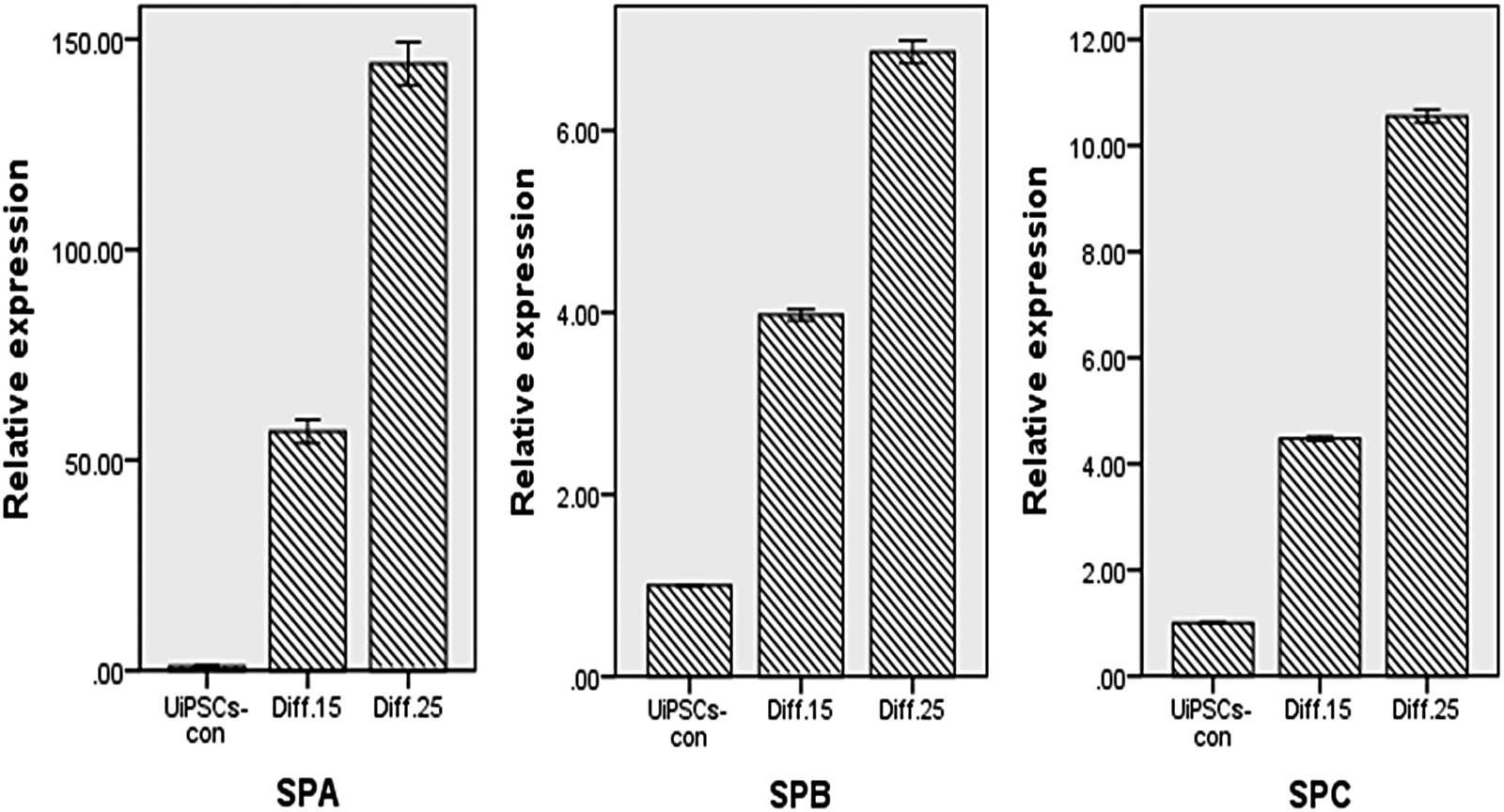
At day 15 (Diff. 15) and 25 (Diff. 25) after differentiation, gene expression of SPA, SPB, and SPC were up-regulated compared with the UiPSCs-con group (p < 0.05.
Discussion
Directed differentiation of human iPSCs into AT II epithelial cells holds great promise for the cellular therapy of lung disease and regenerative medicine, and its clinical application may be fulfilled in the future after sufficient basic research. Yamanaka pioneered iPSCs technology by converting mouse fibroblasts into mouse iPSCs by introducing four retroviral transcription factors (Takahashi and Yamanaka, 2006). Because iPSCs can be derived from adult autologous cells, not only can the ethical issues of the use of embryos be avoided, but also immune rejection of allogenic cells can be bypassed. Different culture methods of obtaining AT II cells from iPSCs have been reported in many investigations (Ghaedi et al., 2013; Huang et al., 2014; Mou et al., 2012; Yan et al., 2014). Huang et al. described a strategy to achieve high yields of AT II epithelial cells by Sendai virus and mRNA transfection of human somatic cells. Yan et al. reported a novel β2-microglobulin–specific insertion targeting strategy using a single nonviral vector for reprogramming human skin fibroblasts and achieving 99% AT II epithelial cells in G418 selected cultures of differentiated iPSCs.
We used a four-stage culture method to induce differentiation of UiPSCs derived from episomal plasmid transfection into AT II epithelial cells in the study. The cells positively expressed SPA, SPB, and SPC at the end of differentiation. The specifically expressed genes of AT II epithelial cells (SPA, SPB, and SPC) increased significantly after differentiation, although the positive percentage was less than the findings of Yan et al. (2014). According to statistical results, the cellular transformation between UiPSCs and AT II cells showed a significant difference (p < 0.05). However, SPA gene expression increased more significantly than SPB and SPC genes in quantitative RT-PCR tests after the differentiation. The reason for the obvious up-regulation of SPA could be that at the end of culture the existing cells contained other cell types, such as bronchial epithelial cells, besides AT II epithelial cells, requiring us to identify more details.
With our four-stage induction protocol, the UiPSCs can be induced successfully into AT II epithelial cells. This protocol provided feasibility for clinical application of UiPSCs-derived AT II cells in cellular therapy of lung diseases caused by dysfunction of AT II cells. The mature AT II epithelial cells have lamellar bodies, which are characteristic structures under the transmission electron microscope. However, it is a limitation that we did not observe this structure in the study. Additionally, we should include a marker that normally is not expressed in AT II epithelial cells, such as Clara cell secretory protein (CCSP). Another limitation is that more time points should be chosen during real-time PCR tests of gene expression to show variation details of specific markers of the AT II epithelial cells.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 31370993).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.