
Abstract
Abstract
The aim of this work was to provide, for the first time, a protocol for isolation and characterization of stem cells from porcine amniotic membrane in view of their potential uses in regenerative medicine. From three samples of allanto-amnion recovered at delivery, the amniotic membrane was stripped from overlying allantois and digested with trypsin and collagenase to isolate epithelial (amniotic epithelial cells [AECs]) and mesenchymal cells, respectively. Proliferation, differentiation, and characterization studies by molecular biology and flow cytometry were performed. Histological examination revealed very few mesenchymal cells in the stromal layer, and a cellular yield of AECs of 10 × 106/gram of digested tissue was achieved. AECs readily attached to plastic culture dishes displaying typical cuboidal morphology and, although their proliferative capacity decreased to the fifth passage, AECs showed a mean doubling time of 24.77 ± 6 h and a mean frequency of one fibroblast colony-forming unit (CFU-F) for every 116.75 plated cells. AECs expressed mesenchymal stem cell (MSC) mRNA markers (CD29, CD166, CD90, CD73, CD117) and pluripotent markers (Nanog and Oct 4), whereas they were negative for CD34 and MHCII. Mesodermic, ectodermic, and endodermic differentiation was confirmed by staining and expression of specific markers. We conclude that porcine amniotic membrane can provide an attractive source of stem cells that may be a useful tool for biomedical research.
Introduction
T
The similarities between pigs and humans make this animal model a valuable tool for evaluation of the safety, feasibility, dosage, engraftment, or toxicity of transferred mesenchymal stem cells (MSCs) (Casal and Haskins, 2006). For practical application of MSCs in regenerative medicine, some aspects of the stem cell reservoir must be considered: large numbers of cells must be harvested in an inexpensive and noninvasive way, without risk to the donor, and these cells must retain a high ability to proliferate and differentiate in vitro (Carlin et al., 2006). The most used and best-characterized source of adult MSCs is bone marrow (BM), but collection of BM requires an invasive procedure. Moreover, the proliferative and differentiation potential of BM-MSCs is limited due to the donor age and the number of in vitro passages (Digirolamo et al., 1999; Guillot et al., 2007; Majors et al., 1997).
Placenta has been proposed as a more suitable candidate for the derivation of MSCs both in human and in veterinary medicine. It includes several tissues, such as umbilical cord blood (Chang et al., 2006; Grewal et al., 2003; Kang et al., 2013; Kern et al., 2006), Wharton jelly (Corradetti et al., 2011; Cremonesi et al., 2008; Hoynowski et al., 2007; Iacono et al., 2012; La Rocca et al., 2009; Lovati et al., 2011; Passeri et al., 2009; Weiss et al., 2008), amniotic membrane (Corradetti et al., 2013; Filioli Uranio et al., 2011; Lange Consiglio et al., 2012; Miki and Strom, 2006; Muttini et al., 2013; Parolini and Soncini, 2004; Rutigliano et al., 2013), and amniotic fluid (Chen et al., 2011; Corradetti et al., 2013; Dev et al., 2012; Gao et al., 2014; Iacono et al., 2012; Lovati et al., 2011; Parolini et al., 2009; Steigman and Fauza, 2007). These tissues could provide a large number of cells without risks to the donor in an inexpensive and noninvasive way, because they are discarded at delivery.
In addition, it is known that human MSCs from extrafetal tissues possess a higher proliferation ability and differentiation potential and a longer telomere length compared to cells derived from adult tissues (Kern et al., 2006; Kögler et al., 2004). Specifically, MSCs isolated from amnion are thought to be in an intermediate stage between embryonic and adult stem cells (De Coppi et al., 2007; Delo et al., 2006; Gucciardo et al., 2009; In't Anker et al., 2004) and are known to prevent rejection of the fetus due to their low immunogenicity and immunomodulatory characteristics (Evangelista et al., 2008; Wang, et al. 2006).
In veterinary medicine, presumptive stem cells have been identified in the epithelial and/or stromal portions of the amnion in canine (Filioli Uranio et al., 2011), equine (Lange Consiglio et al., 2012), bovine (Corradetti et al., 2013), ovine (Muttini et al., 2013), and feline (Rutigliano et al., 2013) species. In this regard, some studies have already demonstrated the clinical relevance of amniotic stem cells. Lange-Consiglio et al. (2012, 2013a, b) isolated equine MSCs from the amniotic membrane as an alternative to BM-MSCs and demonstrated their superior efficacy for the treatment of spontaneous tendon injuries in horses, showing only 4% of relapses of the tendon defect in comparison to BM-MSCs (23.08%).
In swine, MSCs have been isolated from other extrafetal tissues such as Wharton jelly (Carlin et al., 2006), umbilical cord blood (Kumar et al., 2007), and amniotic fluid (Chen et al., 2011). The authors of these studies worked with placental tissues and, although the results confirmed that the isolated cells had stem cell properties, the collection of Wharton jelly and amniotic fluid was invasive, occurring, as it did, from the embryonic disc or from early or mid stage of gestation with sows under general anesthesia. In addition, it was only possible to collect a small amount (2 mL) of umbilical cord blood after delivery (Kumar et al., 2007).
The aim of the study reported here was to isolate, for the first time, presumptive MSCs from porcine amniotic membrane (a usually discarded tissue) at delivery and to characterize these cells in terms of morphology, proliferative and differentiation potential, colony-forming unit (CFU) capability, and expression of stemness markers.
Materials and Methods
Materials
Chemicals were obtained from Sigma-Aldrich Chemical (Milan, Italy) unless otherwise specified, and tissue culture plastic dishes were purchased from Euroclone (Milan, Italy).
Tissue collection
In this study, after approval by the Ethical Committee of the University of Milan and written owner's consent, all procedures were conducted following standard veterinary practice and in accordance with 2010/63 EU directive on animal protection and Italian Law (D.L. No. 116/1992). Allanto-amniotic membranes and mesamnions were collected at delivery from three normal full-term pregnant sows. A mean of four allanto-amniotic membranes and four mesamnions were collected from each sow. Mesamnion, generally found just after delivery adhering to the back of piglet, is where the two chorio-amniotic folds meet and fuse, thus connecting the amniotic membrane with the chorion, as shown in Figure 1A. In pigs, as in cattle, the mesamnion persists until parturition in such a way that offspring are generally born without covering membranes (Hyttel et al., 2011).
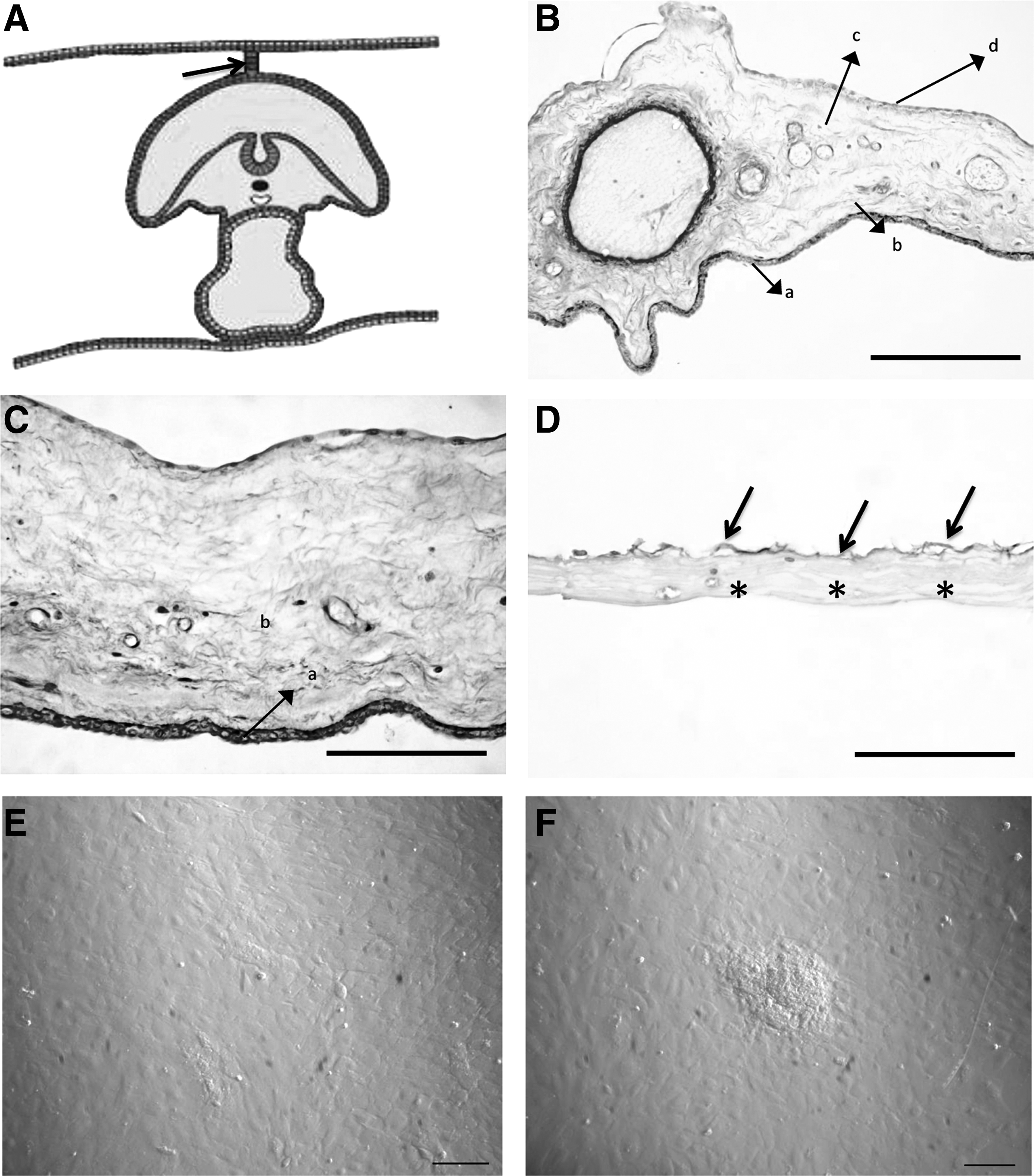
Histology of swine fetal adnexa, PAS reaction. (
Samples for histology were fixed immediately as described in the following section. Other samples were kept at 4°C in calcium- and magnesium-free phosphate-buffered saline (PBS-CMF) (Euroclone), supplemented with 100 U/mL of penicillin, 100 μg/mL streptomycin (Sigma) and 1 μg/mL amphotericin B (Sigma), and processed within 12 h of harvesting.
Histology of allanto-amnion and mesamnion
Paraffin embedding and preparation of histologic sections
For each sow, fragments of the four allanto-amnions and four mesamnions were fixed in 10% formalin for 48–72 h at 4°C. Tissues were dehydrated in a graded series of ethanol and embedded in paraffin. Tissue blocks were cut at 5- to 7-μm thickness using a microtome (Microm International GmbH, Walldorf), and sections were dewaxed and stained for general morphological purposes.
Hematoxylin & Eosin and Periodic Acid Schiff staining
Serial sections were stained either using a routine Hematoxylin & Eosin method for precise anatomical information or with Periodic Acid Schiff (PAS) staining technique to describe better the major tissue types in the specimens. PAS-positive structures stain magenta. Sections were examined under an Olympus BX51 photomicroscope connected to a digital camera and DP-soft (Olympus, Italy) for computer-assisted image acquisition and processing.
Isolation of amniotic mesenchymal and epithelial stem cells
The allanto-amniotic membranes collected were stripped mechanically from the overlying allantois to obtain the thin, transparent amniotic membranes. Then, pools of the amniotic membranes and pools of the mesamnions of each sow (4–5 grams of tissue for every type of membrane) were made. Membranes of each pool were cut into small pieces and digested enzymatically for 9 min at 37°C in a PBS solution containing 2.4 U/mL dispase (Becton Dickinson and Company, Italy).
Cell isolation was performed as previously reported (Lange Consiglio et al., 2012). Briefly, after a resting period of 5–10 min at room temperature in High-Glucose Dulbecco's Modified Eagle Medium (HG-DMEM; Celbio-Corning) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma) and 2 mM
Culture, expansion, and cell count
Soon after isolation, the number of viable cells was counted using the Trypan Blue dye exclusion method (Sigma). Cells were plated at a density of 1 × 105 cell/cm2 in T75 flasks (Euroclone). Cultures were established in HG-DMEM supplemented with 10% FBS, 10 ng/mL epidermal growth factor (EGF; Sigma), 1% penicillin (100 UI/mL) and streptomycin 100 μg/mL, 0.25 μg/mL amphotericin B, and 2 mM
Immunocytoscreen
AECs and AMCs were tested for immunoreactivity against Vimentin (mouse monoclonal, clone Vim V9; Dako, Glostrup, Denmark) and PanCytokeratin (mouse monoclonal, clone A1E; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Cultured AECs and AMCs were fixed in Cytoscreen solution (Hospitex Diagnostics, Milan, Italy) for 48 h and then cytocentrifuged on Superfrost Plus slides (Thermo Scientific, Menzel GmbH & Co., KG, Braunshweig, Germany) and air-dried.
Briefly, for the immunocytochemistry procedure, primary antibodies (anti-Vimentin: 1/100; anti-PanCytokeratin: 1/200) diluted in Tris-buffered saline with Tween 20 (TBST) were applied and incubated overnight at 4°C. Secondary antibodies, conjugated with horseradish peroxidase (HRP), were added and incubated for 30 min at room temperature. The peroxidase reaction was developed for 10 min using diaminobenzidine (DAB), following the manufacturer's instructions (ImpactDAB, Vector Labs Inc., Burlingame, CA, USA) and blocked with deionized water. Cells were considered positive for Vimentin and PanCytokeratin when the presence of intracytoplasmic stain was observed. Negative controls for the target antigens were performed by replacing the primary antibodies with irrelevant antibodies from the host species in which the immunoglobulins were developed [rabbit immunoglobulin (Ig) fraction normal X093 and mouse IgG1 X0931; Dako]. Negative controls in which the primary antibody was replaced with a buffer solution (TBST) alone were also performed.
Proliferation rate and CFU assay
Proliferation rate and CFU assay were determined as previously reported (Lange-Consiglio et al., 2012). Doubling time (DT) was assessed from P1 to P5, CFU assays were performed at P0 plating cells at different densities (100, 250, 500, and 1000 cells/cm2). Colonies formed by 16–20 nucleated cells were counted under a BX71 inverted microscope (Olympus).
Flow cytometry
AECs were analyzed by flow cytometry to determine the percentage of pluripotent- (Oct-4) and mesenchymal- (CD73) associated markers after isolation (P0). Primary antibodies were purchased from Abcam (Cambridge, UK; goat polyclonal antibody Oct-4) and Santa Cruz Biotechnology Inc. (Texas, USA; rabbit polyclonal CD73). Secondary antibodies included donkey anti-goat IgG-AlexaFluor-488 and goat anti-rabbit IgG- AlexaFluor-488 (Abcam, Cambridge, UK). Staining procedure was performed as previously reported (Corradetti et al., 2011). Cells (1 × 106 cells / mL) were labeled with anti-CD73 in PBS with 3% of bovine serum albumin (BSA) (BDH; VWR International Ltd, Poole, UK) for 45 min at room temperature in the dark, followed by washing in cold PBS and final incubation with the secondary antibodies (1:250) for 30 min at room temperature in the dark.
For evaluation of Oct-4, cells (2 × 106 cells / mL) were fixed in 0.01% paraformaldehyde (in PBS) at 4°C for 15 min, washed in 3% BSA (in PBS), and then treated to promote permeability for 10 min at room temperature in 1% Triton-X 100 (in PBS). After incubation, cells were washed twice in ice-cold PBS and analyzed using a Millipore Guava easyCyte™ Single Sample Flow Cytometer. A minimum of 10,000 cells was acquired for each sample and analyzed in the FL1 channel. All analyses were based on control cells incubated with isotype-specific IgGs to establish background signal. Off-line analyses of the flow cytometry standard (FCS) files were performed using Weasel software v.2.5 (http://en.bio-soft.net/other/weasel.html).
In vitro differentiation
To assess their potential to undergo mesodermic (osteogenic and adipogenic), ectodermic (neurogenic), and endodermic (pancreatic) differentiation, AECs were expanded up to P3 and then seeded at a density of 3 × 103 cell/cm2. Cells plated at the density of 1.5 × 103 cells/cm2 were used as control (noninduced cells). When 80% confluence was achieved, the differentiation procedure began. Controls (or the uninduced cells) were cultured for the same time as the differentiation protocol in growth medium.
Osteogenic differentiation
To induce osteogenic differentiation, cells were incubated in HG-DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin B, 2 mM
Adipogenic differentiation
Adipogenic differentiation was performed by stimulating cells with three cycles of induction/maintenance for a 21-day period. The adipogenesis induction medium was composed of HG-DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin B, 2 mM
Neurogenic differentiation
Neurogenic differentiation was performed with a 24-h preinduction in a medium composed of HG-DMEM supplemented with 20% FBS and 1 mM β-mercaptoethanol (Sigma) (Mitchell et al., 2003). Then, neurogenic induction was performed with a medium composed of HG-DMEM supplemented with 2% FBS, 2% dimethyl sulfoxide (Sigma), and 200 μM butylated hydroxyanisole (Sigma). Cells were maintained in this medium for 3 days. Spinal cord was used as positive control.
Pancreatic differentiation
Expanded AECs at P3 were induced to differentiate by a three-step protocol (Gao et al., 2008). In step 1, the cell monolayer was treated for 24 h with HG-DMEM supplemented with 10% FBS and 10−6 M retinoic acid (Sigma); then the medium was changed with HG-DMEM with only 10% FBS for 2 days. In step 2, the cells were detached with 0.25% trypsin–EDTA and seeded in extracellular matrix (ECM) gel- (Euroclone) coated 60-mm plates. The medium was switched to Low-DMEM, supplemented with 10% FBS, 10 mmol/L nicotinamide (Sigma), and 20 ng/mL EGF (Sigma) for 6 days. In step 3, to mature the insulin-producing cells, the low-glucose medium was supplemented with 10% FBS and 10 nmol/L exendin-4 (Sigma) for 6 days. As a control group, the cells were cultured in Low-DMEM containing only 10% FBS.
To confirm differentiation toward the osteogenic, adipogenic, and neurogenic lineages, conventional von Kossa, Oil Red O, and Nissl stainings were performed, respectively, whereas pancreatic differentiation was monitored by observation of three-dimensional, islet-like cell cluster formation. Gene expression of specific differentiation markers was evaluated.
Gene expression analysis (RT-PCR)
Qualitative PCR analysis was performed to evaluate the expression of specific MSC-, pluripotent-, histocompatibility-, and hematopoiesis -associated markers at P0, P1, P3, and P5 and to confirm differentiation. Total RNA was isolated from cells using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. RNA concentration and purity were measured using a NanoDrop Spectrophotometer (NanoDrop ND1000, Wilmington, DE, USA). The cDNA was synthesized from 1 μg of total RNA using the iScript retrotranscription kit (Bio-Rad Laboratories), and a qualitative PCR reaction was performed using the Taq DNA Polymerase recombinant commercial kit (Invitrogen Life Technologies). All amplification reactions were performed using a T1 Thermocycler (Biometra). The conditions of the amplification reaction were: 94°C for 2 min (initial denaturation); 94°C for 30 sec (denaturation); 58–65°C (temperature depending on the melting temperature of primers) for 30 sec (annealing); 72°C for 30 sec (extension); 72°C for 10 min (final elongation). Denaturation, annealing, and extension steps were repeated 32 times.
Primer design
Swine-specific oligonucleotide primers were designed using open source PerlPrimer software v. 1.1.17, based on available NCBI Sus scrofa sequences or on mammalian multialigned sequences. Primers were designed across an exon–exon junction to avoid DNA amplification. Table 1 lists the primers used to characterize AECs and confirm their multidifferentiation potential. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a reference gene.
F, forward; R, reverse; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; BGLAP, osteocalcin; OPN, osteopontin; ADIPQ, adiponectin; LEP, leptin; GFAP, glial fibrillary acidic protein; NES, nestin; INS, insulin; PDX-1, pancreatic and duodenal homeobox 1.
Statistical analysis
Statistical analysis was performed using GraphPad Instat 3.00 for Windows (GraphPad Software, La Jolla, CA, USA). Three replicates for each experiment (growth curve, DT, and CFU) were performed and the results are reported as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) for multiple comparisons by Student–Newman–Keuls multiple comparison tests was used. CFU comparisons among different cell plating densities inside each group were analyzed. Differences were considered statistically significant for p values <0.05.
Results
Histological analysis, cell collection, and morphology
Figure 1, B and C, shows that different contiguous tissues can be distinguished within the allanto-amnion—amniotic epithelial layer, amniotic mesenchymal layer, allantoic mesenchymal layer, and allantoic epithelial layer. The amnion consisted of cuboidal epithelial cells in the epithelial layer (Fig. 1B, Ca) and of stromal cells in the underlying mesenchymal layer (Fig. 1B, Cb). The adjacent allantoic membrane was composed of mesenchymal tissue (Fig. 1B, Cc) covered by the flattened allantoic epithelium (Fig. 1B, Cd). The amniotic and allantoic counterparts were separated mechanically for further analyses. At the time of membrane collection, a thin membrane was harvested from the backs of piglets. On the basis of histologic analysis and of its positioning, this membrane was thought to be the mesamnion (Fig. 1D). Both the allanto-amnion and the mesamnion showed a paucity of cells in their mesenchymal components. Actually, no cells were obtained from the stromal layer of the amnion or the mesamnion, and only the amnion epithelial layer proved to be an efficient source of cells to be plated and maintained in culture (AECs). A concentration of 10 × 106 cells/gram of digested tissue was obtained after digestion and viability of the isolated cells was higher than 95%. AECs showed a typical polygonal morphology (Fig. 1E). Cell colonies, observed since the first culture steps, demonstrated an ability to organize into three-dimensional structures and to form clusters (Fig. 1F).
Immunocytoscreen
AECs were strongly positive for PanCytokeratin and negative for Vimentin (Fig. 2A, B).
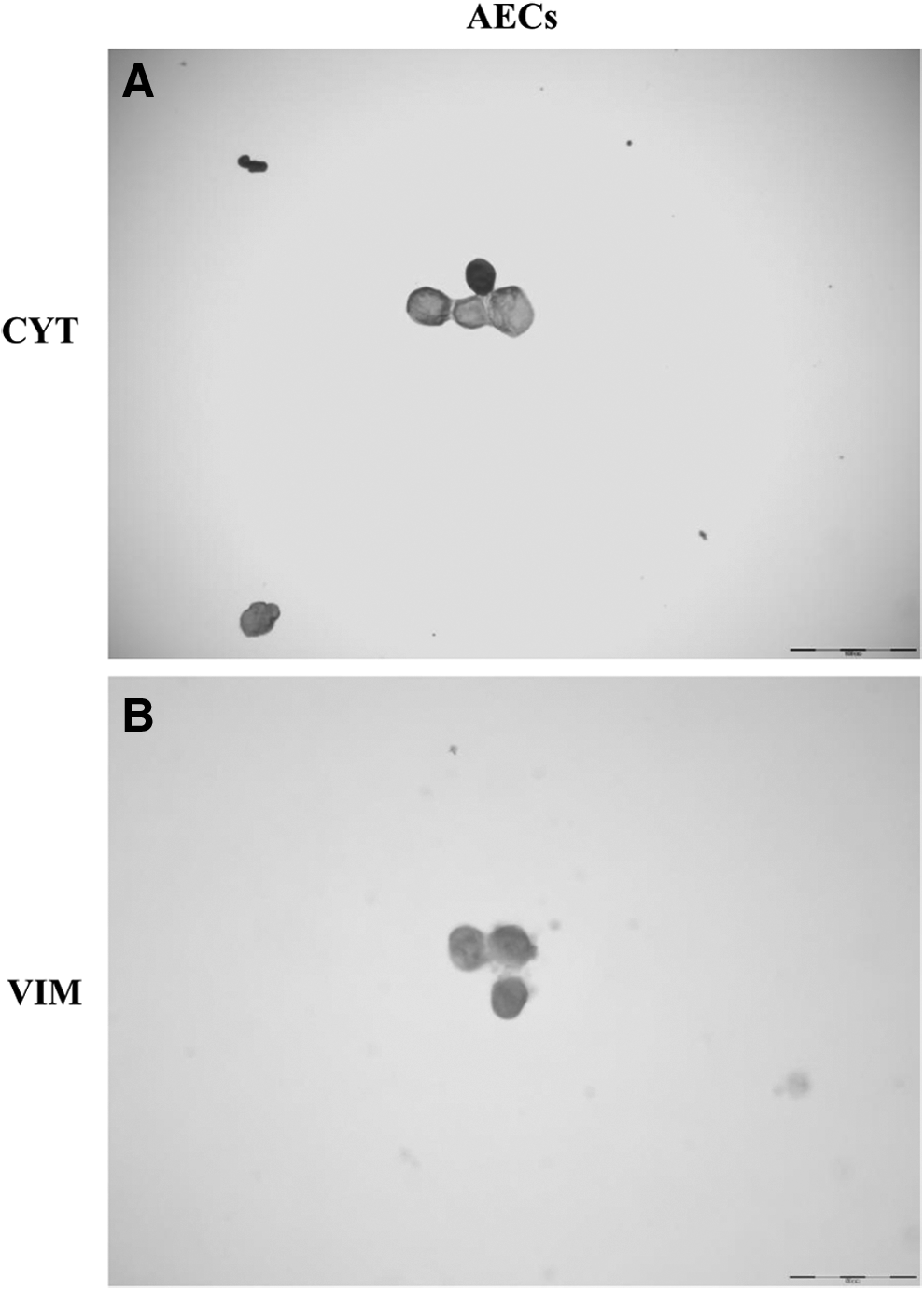
Immunocytoscreen results. Immunolocalization of cytokeratins (CYT) and vimentin (VIM) in AECs (
Proliferation analysis
Figure 3A shows the growth curve for AECs at P1, P3, and P5. AECs showed a growth curve, with an initial lag phase of few hours and a further intense log phase of 2–12 days at P1 and P3. At P5, AECs showed a slow lag phase (about 4 days) and a less intensive log phase. Statistically significant differences (p < 0.05) in DT were identified between the passages studied (see asterisks in Fig. 3B). After P1, the proliferation rate increased; indeed, the values decreased at P2 and remained constant until P3. The mean DT value was 24.77 ± 6 h. A statistically significant increase (p < 0.05) in fibroblast CFUs (CFU-F) frequency was observed, reflecting the increase of plating density (Table 2).
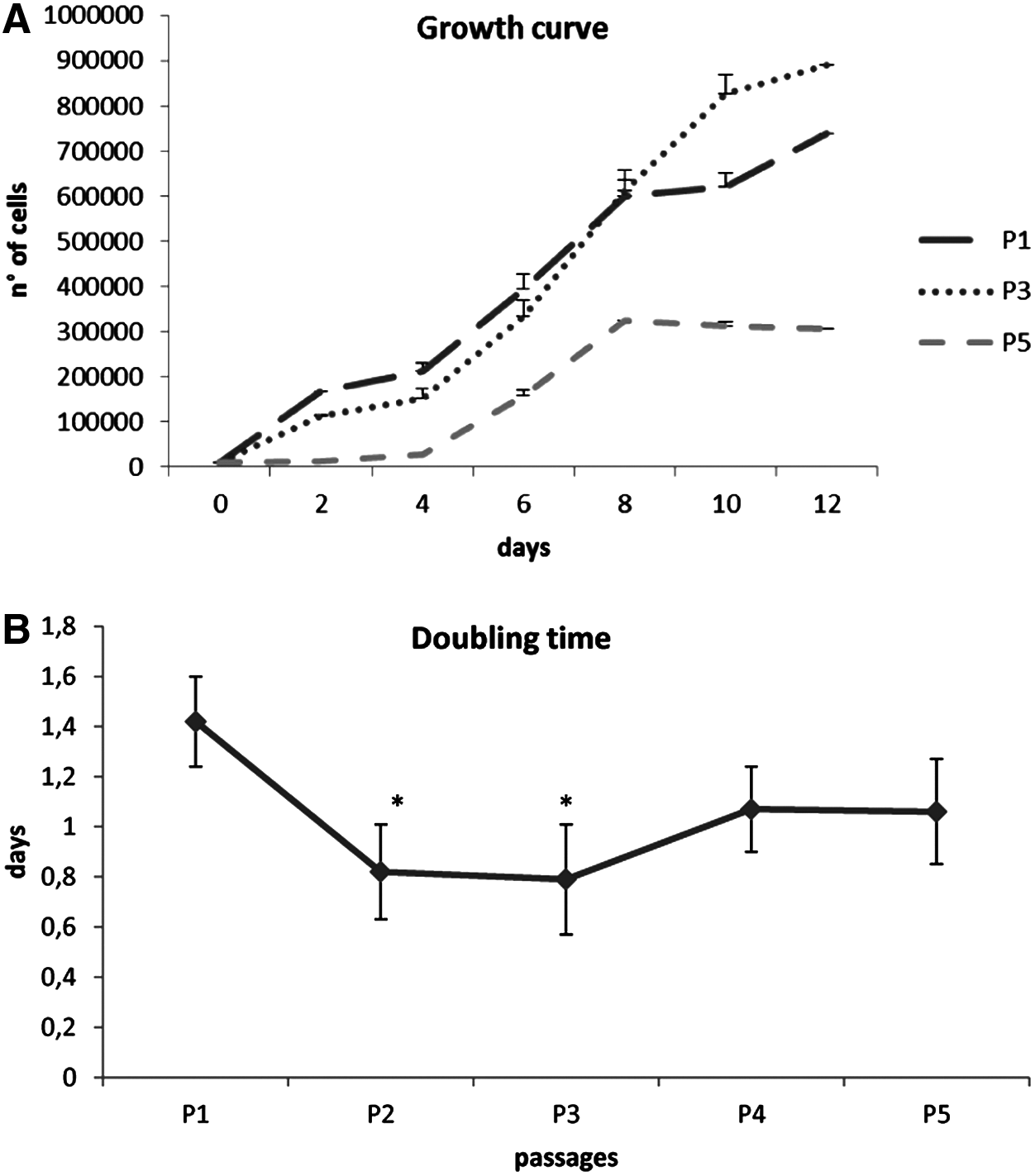
Proliferative potential. (
Different superscripts (a, b) indicate statistically different comparisons (p < 0.05) between cell densities in each group.
CFU-F, fibroblast colony-forming units.
Flow cytometry analysis
Flow cytometry analysis was assessed to evaluate the homogeneity of the cell population and to identify the subset of pluripotent or mesenchymal cells. The cell population tested resulted homogeneously composed of CD73- and Oct-4–positive cells, as reported in Figure 4.
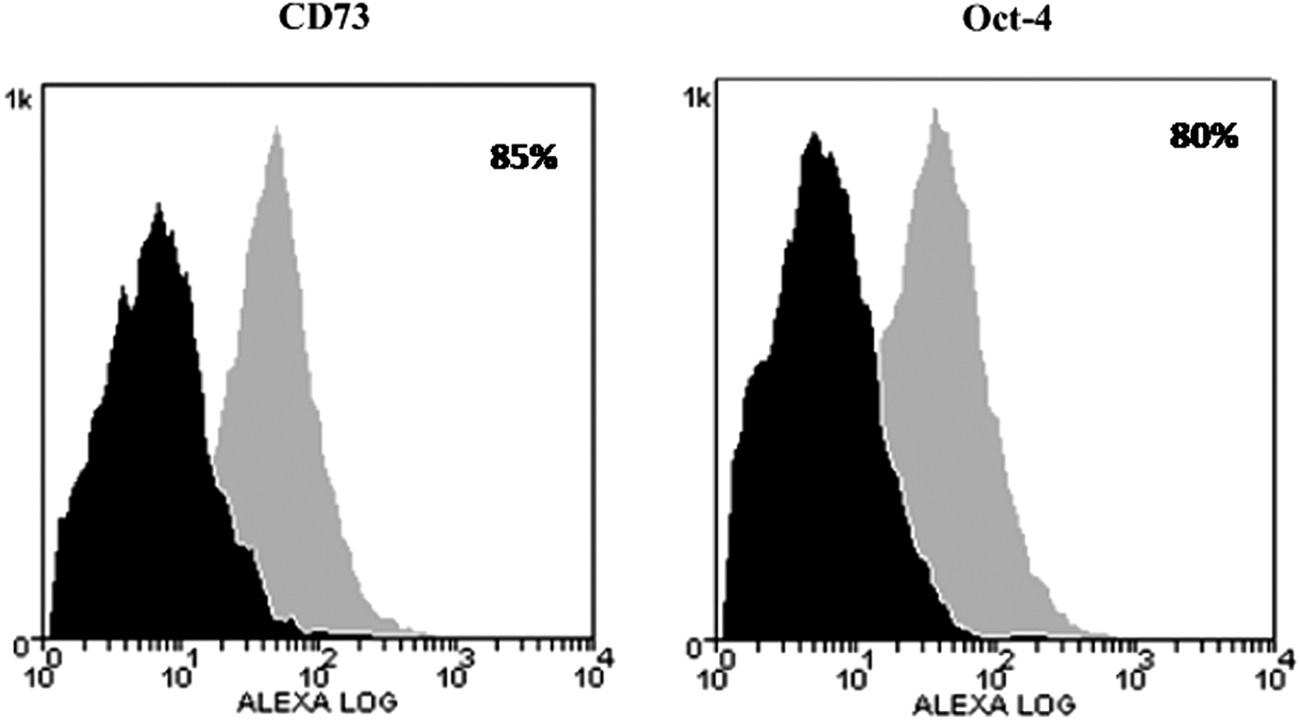
Flow cytometry analysis of antigen expression with Alexafluor-488 labeled antibodies: Oct-4 and CD73. Histograms represent relative number of cells vs. fluorescence intensity (FL1). Black histograms indicate background fluorescence intensity of cells labeled with isotype control antibodies only; grey histograms show positivity to the studied antibodies.
In vitro differentiation
At P3, AECs were shown to be plastic and able to undergo osteogenic, adipogenic, neurogenic, and pancreatic differentiation. After 21 days of induction, the osteogenic differentiation of AECs was confirmed by Von Kossa staining, which highlighted calcium precipitates. Moreover, cells modified their morphology and increased their size. The control sample did not stain and did not develop a mineralized matrix. The osteogenic induction was further verified through RT-PCR analysis of osteogenesis-associated markers expression—osteocalcin (BGLAP) and osteopontin (OPN). Induced cells expressed OPN and BGLAP, whereas control cells did not. Bone was used as positive control for the expression of osteogenic markers (Fig. 5A).
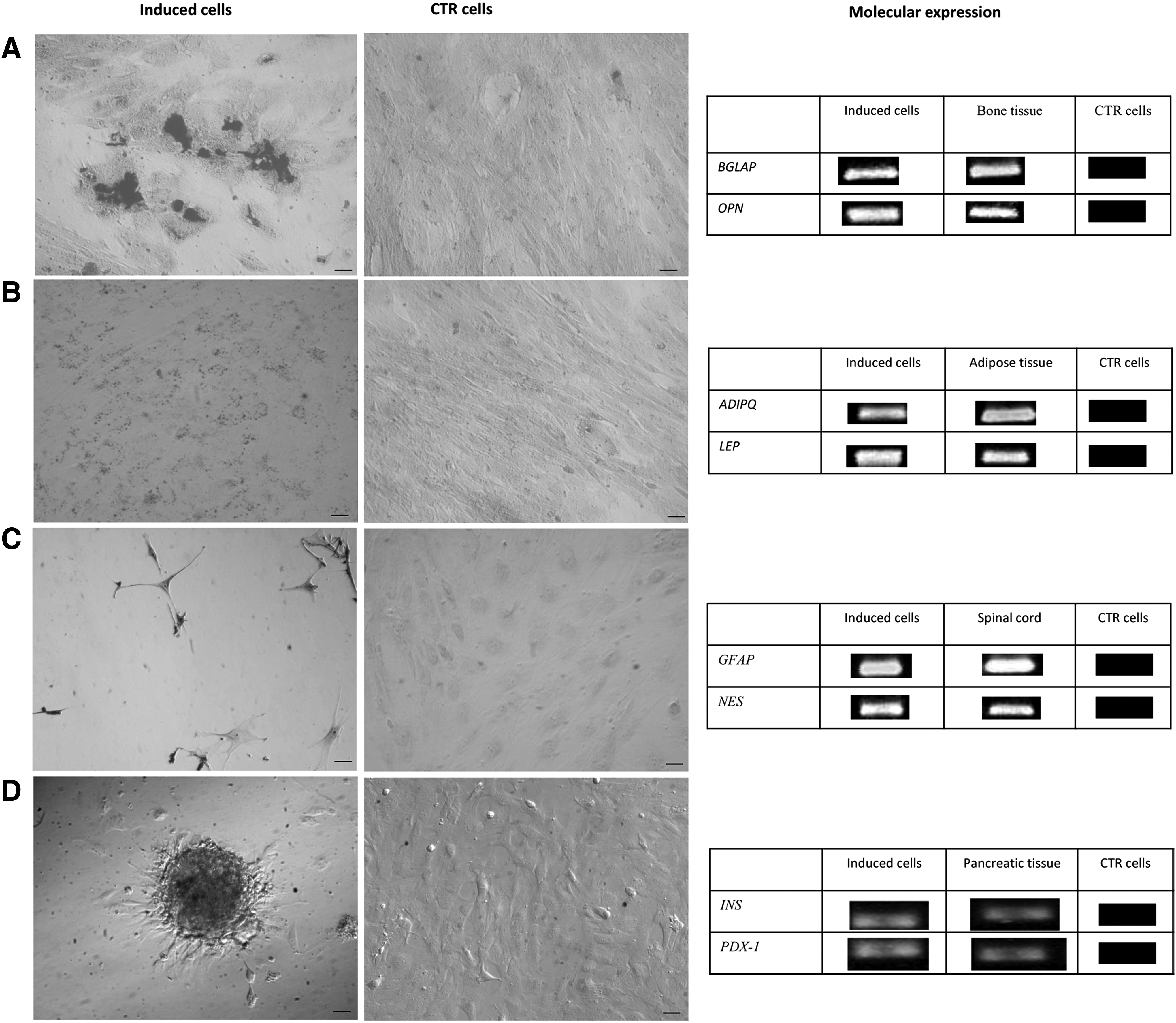
Differentiation potential. (
After 3 weeks of induction, AECs demonstrated differentiation ability. Indeed, after adipogenic induction, the Red Oil O staining showed intracellular lipid vacuoles that were not present in control AECs maintained in standard medium. RT-PCR for the expression of adipogenic markers such as adiponectin (ADIPQ) and leptin (LEP) confirmed adipogenic induction. Adipose tissue was used as positive control (Fig. 5B).
After 3 days of neurogenic induction, stimulated cells acquired a typical neuronal morphology and stained positively for Nissl, whereas uninduced cells did not. The analysis of neurogenic expression markers, such as the glial fibrillary acidic protein (GFAP) and nestin (NES) confirmed the neurogenic induction. Spinal cord was used as positive control (Fig. 5C).
After 15 days of induction, AECs changed their typical epithelial shape and gradually organized large three-dimensional colonies (Fig. 5D), reminiscent of typical pancreatic islets in vitro. Expression of insulin and pancreatic and duodenal homeobox 1 (PDX-1) mRNA transcripts were detected by RT-PCR.
Evaluation of marker expression by RT-PCR
To characterize amniotic stem cells, RT-PCR analysis was performed on cells at different passages (P0, P1, P3, P5). As shown in Figure 6, cells expressed mesenchymal-associated markers (CD166, CD117, CD29, CD90, CD73), embryonic-associated markers (Nanog, Oct-4, Sox2), and the immunological marker MHC-I. They did not express MHC-II nor the hematopoietic marker CD34 from P0 to P5. Of particular interest is the CD117 expression that decreased from P0 to P5,

RT-PCR analysis. Specific gene expression in AECs from P0 to P5 of mesenchymal (CD166, CD117, CD29, CD90, CD73), pluripotent (Nanog, Oct-4, Sox2), hematopoietic (CD34) and histocompatibility (MHC-I and MHC-II) mRNA markers. GAPDH was used as reference gene.
Discussion
The advantage of using amniotic membrane for production of stem/progenitor cells in human and animal species is the availability of the large amount of these tissues and the noninvasive collection procedure at birth. Because the amniotic membrane is an extrafetal tissue, we conjectured that amniotic cells with the potential to be primitive mesenchymal cell types might also exist in full-term porcine amnion. Moreover, considering the similarity between pigs and humans, the chance to characterize porcine stem cells from this source could be helpful in experimental cell-based therapies.
The aim of this study was to isolate and characterize presumptive stem cells from the porcine amniotic membrane, suggested in several studies to be an optimal source of pluripotent/multipotent stem cells in a variety of veterinary species (Corradetti et al., 2013; Filioli Uranio et al., 2011; Lange Consiglio et al., 2012; Muttini et al., 2013; Rutigliano et al., 2013). Usually, two cell types are harvested from this tissue—amniotic epithelial and mesenchymal cells. In contrast to findings in other species, in the present study we were able to isolate only AECs from the amniotic membrane; the mesenchymal layer of amniotic membrane contained few stromal cells. Concerning the mesamnion, few epithelial cells and no stromal cells could be retrieved. For this reason, only amniotic AECs were expanded, characterized, and differentiated. After digestion, large numbers of AECs with greater than 95% viability (optimal value in terms of plating efficiency and cellular growth) were obtained. Positivity to cytokeratin and negativity to vimentin confirmed that isolated cells were epithelial cells.
When cultured, AECs demonstrated strong adherence to plastic dishes and developed epithelial polygonal morphology over time. Adherence is a fundamental property of cultured stem cells (Dominici et al., 2006). When AECs were kept in culture, small cell clusters or spheroid structures developed, showing that these cells lacked contact-inhibited cell growth and continued to proliferate after reaching 100% surface confluence, forming aggregates overlying the monolayer of confluent cells. Other investigators have reported that the monolayer of amniotic cells may support the growth and lack of differentiation in amniotic cells forming the spheroid structures, possibly playing the role of an autologous feeder layer and providing secreted factors (Miki et al., 2010).
The proliferation study showed that AECs reached high plating efficiency and had a high proliferation rate in vitro until P3, when AECs showed a growth curve with a lag phase of few hours and an intensive log phase of 12 days. Moreover, the DT value at P3 was 19 h. These data are in agreement with those obtained by other researchers who reported a high proliferation rate of human (Miki et al., 2010; Soncini et al., 2007), equine (Lange-Consiglio et al., 2012), bovine (Corradetti et al., 2013), and feline (Rutigliano et al., 2013) amniotic cells. After this period, the proliferation rate decreased.
During the different subcultures, the time required to remove adherent cells from a culture surface using trypsin increased reaching a maximum of about 20 min. It is likely that prolonged treatment with trypsin degrades membrane proteins, causing alterations in the cells' ability to adhere to the plate. Indeed, at P5 the lag phase was 4 days. For this reason, in this preliminary study, cell expansion could not be continued beyond P5. This problem, which can be circumvented by use of other techniques for cell detachment (for example using scrapers), does not affect the potential therapeutic use of these cells because in regenerative medicine cells are usually used at P3. Indeed, this passage is considered the best passage to obtain homogeneity of cells and to prevent aging in the plate that would lead to epigenetic changes in gene expression related to in vitro culture (Lange-Consiglio et al., 2012).
Swine AECs also showed the ability to produce clones and revealed the typical expression pattern expected for cultured stem cells (Dominici et al., 2006) when analyzed by RT-PCR and flow cytometry. It has been demonstrated in both human (Manuelpillai et al., 2011; Marongiu et al., 2010; Miki et al., 2010; Soncini et al., 2007) and animal MSCs (Chen et al., 2011; Corradetti et al., 2013; Lange-Consiglio et al., 2012; Lovati et al., 2011; Iacono et al., 2012) that pluripotent- and mesenchymal-associated markers are expressed. In our results, the same expression pattern occurred, with the exception of CD117, which decreased substantially from P0 to P5, as reported in some other studies (Meirelles et al., 2003, 2006). The isolated cells are epithelial cells, but they expressed MSC markers. Bilic et al. (2008) reported that human AECs had an antigenic expression profile characteristic of culture-expanded MSCs and could co-express epithelial and mesenchymal cell markers (Sakuragawa et al., 2004).
In fact, it has been reported that the amnion-derived cells have not completely differentiated into the epithelial or mesenchymal phenotype; another explanation is that the epithelial–mesenchymal transition may occur in the amniotic membrane (Bilic et al., 2008; Sakuragawa et al., 2004). Expression of the hematopoietic marker CD34 was not found at any passage. This result was expected, and it confirmed that isolated cells do not belong to a hematopoietic lineage.
To assess the usefulness of porcine AECs for cell therapy, expression of markers related to cell immunogenicity, including MHC-I and MHC-II, was also evaluated. At each passage studied, AECs were negative for MHC-II and positive for MHC-I, consistent with previous reports (Corradetti et al., 2013; Lange-Consiglio et al., 2012), thus reinforcing the role of the amniotic membrane as an allogenic source for cell-based therapies in pigs.
Differentiation data confirmed the high plasticity of these cells that can differentiate into mesodermic (osteogenic and adipogenic), ectodermic (neurogenic), and endodermic lineages. After 21 days of induction, mineral deposits were confirmed by Von Kossa staining and by the expression of BGLAP and OPN. These results suggest that these cells reached a mature osteogenic differentiation, as BGLAP and OPN genes are associated to the matrix maturation and mineralization phases (Raouf and Seth, 2000). Stimulated AECs were also able to undergo adipogenic differentiation confirmed by expression of LEP and ADIPQ (regarded as intermediate and late markers of adipocyte differentiation). Moreover, AECs acquired a neuron-like morphology and expressed specific neuronal markers expression (GFAP and NES) after in vitro induction. The morphological changes and the marker expression confirmed that the cell population comprised neuronal precursor stem cells (Lendahl et al., 1990), as observed for amniotic-fluid derived MSCs in the same species (Zheng et al., 2010). Regarding the pancreatic differentiation, morphological and molecular studies suggest that the differentiation occurred, but further study will be needed to confirm the ability of these induced cells to secrete insulin.
This work allowed the isolation, characterization, and differentiation of stem cells derived from the epithelial layer of the amniotic membrane in pigs. Altogether, our findings suggest that porcine amniotic membrane is a reliable source of presumptive AECs, displaying intermediate features between adult and embryonic stem cells, and could have a wide clinical application because of their low immunogenicity and high differentiation potential. Only epithelial cells were isolated here, but the amniotic epithelium layer, while originating from the trophectoderm as with other parts of fetal membranes, has the peculiarity of being continuous with the epiblast (Vejlsted, 2010). For this reason, it may probably preserve some of the characteristics of the epiblast (Miki et al., 2006), such as Nanog and Oct4 expression.
Conclusion
The advantages of this study are the source of cells (amniotic membrane) and the pig as an animal model. Collection of the amnion, in comparison to embryonic and adult tissues, does not pose ethical issues, and its retrieval at term is a noninvasive procedure. One of the most important criteria when choosing an animal model is its relevance to human conditions. Over the years, a huge amount of information, mostly conducted on standard laboratory animals such as rodents, has been generated. When new therapies for human use are designed using this data, they often fail, particularly if the metabolism of xenobiotics of interest is different.
Aside from nonhuman primates, the pig is the species that shares the closest evolutionary resemblance to humans (Verma et al., 2011); thus, its human-like physiology means that data obtained in this species are highly relevant for human-related therapeutic research. In addition to the potential use of porcine amniotic cells as candidates in auto/allo/xenogenic transplantation, these AECs are widely available and could also be used to evaluate the safety and efficacy of cell therapies.
Footnotes
Acknowledgment
This study was supported by grants from Università degli Studi di Milano and Università Politecnica delle Marche, Ancona, Italy.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.