
Abstract
Abstract
Recent studies have revealed the presence of a mesenchymal stem cell (MSC) population in human and in gilt granulosa cells (GCs), thus increasing the interest in identifying the same population in the bovine species. We first isolated GCs by scraping from bovine preovulatory follicles and then tested several different media to define the ideal conditions to select granulosa-derived stem cells. Although expressing MSC-associated markers, none of the media tested proven to be efficient in selecting MSC-like cells that were able to differentiate into mesodermic or ectodermic lineages. We performed another experimental approach exposing cells to a chemical stress, such as lowering of pH, as a system to select a more plastic population. Following the treatment, granulosa-specific granulose markers [follicle-stimulating hormone receptor (FSHR), follistatin (FST), and leukemia inhibitory factor receptor (LIFR)] were lost in bovine GCs, whereas an increase in multi- (CD29, CD44, CD73) and pluripotent (Oct-4 and c-Myc) genes was noticed. The stress allowed up-regulation of tumor necrosis factor-α and interleukin-1β expression and the dedifferentiation of GCs, which was demonstrated by differentiation studies. Indeed, pH-treated cells were able to differentiate into the mesodermic and ectodermic lineages, thus suggesting that the chemical stress allows for the selection of cells that are more prone to adjust and respond to the environmental changes.
Introduction
A
On the basis of these considerations, the interest of clinicians in finding alternative sources of multipotent cells has increased over the past years. MSCs from the fetal adnexa have been reported as a potential tool to overcome some of these limitations, opening up new prospects for the development of regenerative medicine (Lange-Consiglio et al., 2012). Also in bovine species, MSCs have been obtained from umbilical cord blood (Raoufi et al. 2011) and amnion and amniotic fluid (Corradetti et al., 2013). Recently, other research groups have suggested granulosa cells (GCs) as a promising adult source of MSCs in humans (Kossowska-Tomaszczuk et al., 2009) and in gilts (Mattioli et al., 2012). The investigation of Kossowska-Tomaszczuk et al. (2009) refers to luteinizing GCs recovered from preovulatory follicles of patients treated for oocyte retrieval. Mattioli et al. (2012) compared GCs retrieved from growing follicles before the preovulatory gonadotropin surge, and hence before luteinization (e.g., in a condition of active proliferation and before the final differentiation) with those obtained from preovulatory follicles. In both studies, cells expressed MSC-associated markers and were able to differentiate into different cell types not present within the follicles, thus suggesting their potential use in cell-based therapy. Moreover, Mattioli et al. (2012) demonstrated that luteinizing GCs have more efficient osteogenic potential compared to GCs isolated from growing follicles.
GCs represent a readily available source from which to isolate cells easily, relying on the well-established techniques for oocyte retrieval used in assisted reproduction. This has increased our interest in identifying a MSC-like population in the bovine species, considering that ovaries can be easily collected in slaughterhouses due to the absence of their commercial value. In this context, the present study was designed to define a suitable protocol to select GCs with MSC-associated features in terms of morphology, specific markers, and differentiative potential from bovine preovulatory follicles.
Material and Methods
Samples were collected from bovine species slaughtered in a slaughterhouse (INALCA, Ospedaletto Lodigiano, Lodi, Italy) under legal regulations. Chemicals were obtained from Sigma-Aldrich Chemical (Milan, Italy) unless otherwise specified, and tissue culture plastic dishes were purchased from Euroclone (Milan, Italy).
Collection of ovaries and cells isolation
Bovine GCs employed in this study were isolated from ovaries collected from slaughterhouses. The age, genealogy, physiological status, and race were unknown for the animals. The transport of the gonads to the laboratory occurred in a portable thermos maintained at a temperature of 30°C in physiological saline solution (0.9% NaCl) supplemented with kanamycin (150 mg/liter). This temperature was maintained at a constant level from the beginning of ovary collection and during the time needed to reach the laboratory. In the laboratory, the ovaries were washed repeatedly in physiological saline solution supplemented with antibiotics; they were maintained in a thermos for the period of sampling to avoid thermal shock.
GCs were isolated from the bovine ovaries by scraping (Mattioli et al., 2012). Briefly, follicles with a diameter between 0.8 and 1.2 cm were opened using a scalpel blade, and GCs were gently scraped away from the internal face of the follicle wall. The material collected by scraping was deposited into 50-mL tubes containing Tissue Culture Medium (TCM)-HEPES (Sigma) supplemented with 1 mM pyruvic acidic (Sigma), 2.2 grams/liter of sodium bicarbonate (Sigma), 100× penicillin/streptomycin (Sigma), and 10% fetal calf serum (FCS; Sigma). The tubes were left at room temperature until the oocytes sedimented under gravity to the bottom of the tubes. The deposited portion was aspirated and plated on a gridded Petri plate (100 × 15 mm) to select and discharge oocytes. This procedure was performed using a stereomicroscope (model SZX-ILLK200, Olympus) with a 40× enlargement equipped with a heating plate set at 38°C. The GCs were pooled, washed in TCM-HEPES through two successive centrifugations (200 × g for 5 min at room temperature), counted in a Burker chamber, and used in the following experiments.
Experimental design
GCs were collected from bovine preovulatory follicles by scraping, cultured in different media or exposed to acid conditions (pH 5.7), induced to differentiate to mesodermic and ectodermic lineages, and analyzed by qualitative and quantitative PCR. All experiments were performed in triplicate.
GC isolation and culture
At first, GCs isolated by scraping were randomly allocated to five different culture media with the basic medium (MB) consisting of High-Glucose Dulbecco's Modified Eagle Medium (HG-DMEM) supplemented with 10% FCS, 100 IU/mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin B, and 2 mM
This protocol was modified in our study because BME was maintained only for passage 1 (P1) and then removed from the culture medium. For each condition, cells cultures were established in an atmosphere of 5% CO2 and 90% humidity and at a temperature of 38.5°C. Medium was replaced 72 h after isolation to remove nonadherent cells. Adherent cells were detached with 0.05% trypsin-EDTA (EuroClone) just prior to reaching confluence (80%) and then reseeded for culture maintenance. Cells were expanded for three passages, at which point they were analyzed for the expression of specific markers by qualitative and quantitative PCR and were tested for multidifferentiative potential.
In a second step, GCs isolated by scraping were divided into two portions: One was cryopreserved and used as a control [day 0 (d0)] and the second one was exposed to acidic conditions (pH 5.7) and was observed constantly for 7 days to assess the viability and morphological changes (d7). The acidic treatment was performed for 25 min at pH 5.7 and at 37°C (Hjelmeland et al., 2011). After that, cells were centrifuged and the pellet was resuspended in DMEM/F12 medium supplemented with LIF and B27. The cellular suspension was seeded at a density of 1 × 105 cell/cm2 in T25 flasks.
In vitro differentiation
GCs from each condition were seeded at the density of 3 × 103 cells/cm2 for differentiation in adipogenic, osteogenic, and neurogenic lineages. Cells plated at a density of 1.5 × 103 cells/cm2 were used as control. For the first 3–4 days, the cells were incubated with basic medium to allow adhesion; at 60–70% of confluence, they were induced to differentiate.
Osteogenic differentiation
This was assessed by incubating cells for up to 3 weeks at 38.5°C under 5% CO2 in medium composed of HG-DMEM medium supplemented with 10% FBS, 100 IU/mL penicillin, 100 mg/mL streptomycin, 0.25 mg/mL amphotericin B, 2 mM
Adipogenic differentiation
Near-confluent cells were cultured through three cycles of induction/maintenance to stimulate adipogenic differentiation. Each cycle consisted of feeding the GCs for 3 days with supplemented adipogenesis induction medium, followed by culture for another 3 days (38.5°C, 5% CO2) in supplemented adipogenic maintenance medium. The induction medium consisted of HG-DMEM supplemented with 10% FCS, 100 IU/mL penicillin, 100 mg/mL streptomycin, 0.25 mg/mL amphotericin B, 2 mM
Neurogenic induction
This was performed by culturing cells for 24 h in preinduction medium consisting of HG-DMEM, 20% FCS, and 1 mM BME (Mitchell et al., 2003; Seo et al., 2009). Neural induction was performed by switching to a medium composed of DMEM plus 2% FCS, 2% dimethylsulfoxide, and 200 mM butylated hydroxyanisole for 3 days (Woodbury et al., 2000). Noninduced control cells were cultured for the same time period in standard medium. Neurogenic differentiation was demonstrated by conventional Nissl staining (0.1% Cresyl Violet solution) to detect increase of Nissl bodies.
Molecular biology study
Qualitative PCR analysis was performed to evaluate the expression of specific granulosa-, MSC-, pluripotent-, histocompatibility- and hematopoiesis-associated markers to confirm the differentiation that occurred and the stress induced by the acidic treatment. RNA was isolated using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) according to the protocol indicated by the manufacturer. RNA concentration and purity were measured using a NanoDrop spectrophotometer (NanoDrop ND1000, Wilmington, DE, USA). Complementary DNA (cDNA) was synthesized from 300 ng of total RNA using the PrimeScript RT reagent kit (Takara Bio, Otsu-Shi, Japan). Gene expression evaluation was performed using specific sequences. Bovine-specific oligonucleotide primers were designed using open source PerlPrimer software (v. 1.1.17), based on available National Center for Biotechnology Information (NCBI) Bos taurus sequences or on mammalian multialigned sequences. Primers were designed across an exon–exon junction to avoid DNA amplification. Primers sequences and characteristics are reported in Table 1. The bovine glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) was employed as a reference gene in each sample to standardize the results by eliminating variation in messenger RNA (mRNA) and cDNA quantity and quality.
MHC I, major histocompatibility complex class I; MHC II, major histocompatibility complex class II; FSHR, follicle-stimulating hormone receptor; LIFR, leukemia inhibitory factor receptor; LEP, leptin; PPARγ, peroxisome proliferator-activated receptor gamma; BGLAP, bone gamma-carboxyglutamate protein; SPP1, osteopontin; SPARC, secreted protein acidic and rich in cysteine; GFAP, glial fibrillary acidic protein; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Conventional qualitative PCR was performed using 1 μL of the cDNA obtained in a 25-μL final volume with recombinant Taq DNA polymerase (Invitrogen, Life Technologies, Monza, Italy). Amplified PCR products were electrophoresed on a 1.8% agarose gel with ethidium bromide.
For quantitative PCR, one single representative gene per set of markers (CD73, FSHR, and Oct-4) was chosen to evaluate the selection efficiency of any culture condition used in this study. In addition, the expression of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) was evaluated to confirm cellular stress induced by acidic treatment. Analyses were carried out with the SYBR (a fluorescent intercalating agent, able to bind the DNA in double-stranded conformation) method, in the MyiQ™ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA, USA). Triplicate PCR reactions were carried out for each analyzed sample. Reactions were set on a strip in a final volume of 25 μL by mixing, for each sample, 1 μL of cDNA, 12.5 μL of 2× concentrated SYBR® Select Master Mix (Applied Biosystems, Foster City, CA, USA), 1 μM forward primer, 1 μM of reverse primer, and Milli-Q water.
Immunocytochemical characterization of Oct-4
To test the expression of the Oct-4 marker, primary antibody was purchased from Abcam (Cambridge, MA, USA), whereas Alexa Fluor 488–conjugated secondary antibody and goat serum were from Life Technologies (Carlsbad, CA, USA). All products were used following the manufacturer's instructions.
For immunostaining, GCs after isolation (control cell) and GCs cultured in MB + LIF were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and washed three times in phosphate-buffered saline (PBS). After fixation, cells were permeabilized for 15 min at room temperature in 0.25% Triton-X 100 diluted in PBS and washed three times in PBS. After washing, cells were blocked using 10% goat serum in PBS for 30 min at room temperature. Cells were incubated with primary antibodies (1:200 dilution) overnight at 4°C. After washing three times, cells were incubated with secondary antibodies conjugated to Alexa Fluor 488 (1:250 dilution) for 1 h. Finally, for nuclear staining, cells were incubated for 15 min with Hoechst 33342 (1 mg/mL; Sigma) diluted 1:100 in PBS. Images were captured on a BX51 microscope (Olympus, Japan). A negative control was performed without the primary antibodies. The positive control was carried out on bovine in vitro embryos produced routinely in our laboratory as described by Lange-Consiglio et al. (2010).
Statistical analysis
For quantitative PCR data, nonparametric tests were used. The Mann–Whitney U-test was employed to compare two groups (treated vs. untreated). Results were considered statistically significant if p < 0.05.
Results
GCs yield and morphology
From each ovary, about 2 million GCs were isolated with an 80% viability evaluated by Trypan Blue dye exclusion. Cells were plated and selected on the basis of their ability to adhere to plastic. Observation under microscopy revealed the presence of cells with epithelial morphology when cultured in MB (Fig. 1A), with atypical morphology in LIF + BME (Fig. 1B), whereas they displayed fibroblast-like morphology when cultured under other conditions (Fig. 1C–E). After pH treatment, in the next 7 days of culture, cells displayed morphological changes compared to initial epithelial morphology and a progressive vacuolization (Fig. 2). After acidic treatment, the number of viable cells immediately decreased 50%. During the following 7 days of culture, the viability of the remaining cells was further reduced 40%.

Morphology of cells cultured in different culture conditions: (

Morphology of cells isolated from bovine preovulatory follicles and exposed to a chemical stress (d7) display a progressive vacuolization. Scale bar, 20 μm; magnification, 20×.
Molecular analysis of GCs
Table 2 shows expression of GCs studied in different culture conditions. These cells expressed mesenchymal (CD29, CD44, CD166, and CD73) and pluripotent (Oct-4 and c-Myc) markers and lacked CD34 marker expression. For these markers, no differences induced by the culture conditions were observed compared to P0, except for CD166, whose expression was not detected at P0. On the other hand, changes in the granulosa-associated markers were detected in the different culture conditions. Specifically, follicle-stimulating hormone receptor (FSHR) was expressed only at P1 when cells were cultured in MB, MB + EGF, MB + LIF, and MB + LIF + BME. Cells cultured in 2% MB expressed FSHR over the passages studied. Follistatin (FST) expression was observed in all of the conditions and passages, but disappeared at P3 in MB + LIF + BME.
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; FSHR, follicle-stimulating hormone receptor; FST, follistatin; MB, basic medium; EGF, epidermal growth factor; LIF, leukemia inhibitory factor; BME, β-mercaptoethanol.
qPCR results highlighted differences in the expression of FSHR between cells cultured in different conditions (Fig. 3). In MB + LIF and MB + EGF, FSHR expression was found about 10 times less compared to the baseline (P0). Oct-4 expression reached the maximum expression in MB + LIF medium (157.8 ± 65.07) and the minimum expression in 2% MB and MB (0.83 ± 0.38 and 1.73 ± 0.27, respectively). A relatively high value was recorded also in MB + EGF and MB + LIF + BME (4.1 ± 1.1 and 39.7 ± 13.6). The expression level of CD73 was found higher in MB (49.7 ± 23.19) and MB + LIF + BME (29.75 ± 19.87). The lowest expression of CD73 was registered in 2% MB, showing a 1.37- (± 0.44) fold increase.
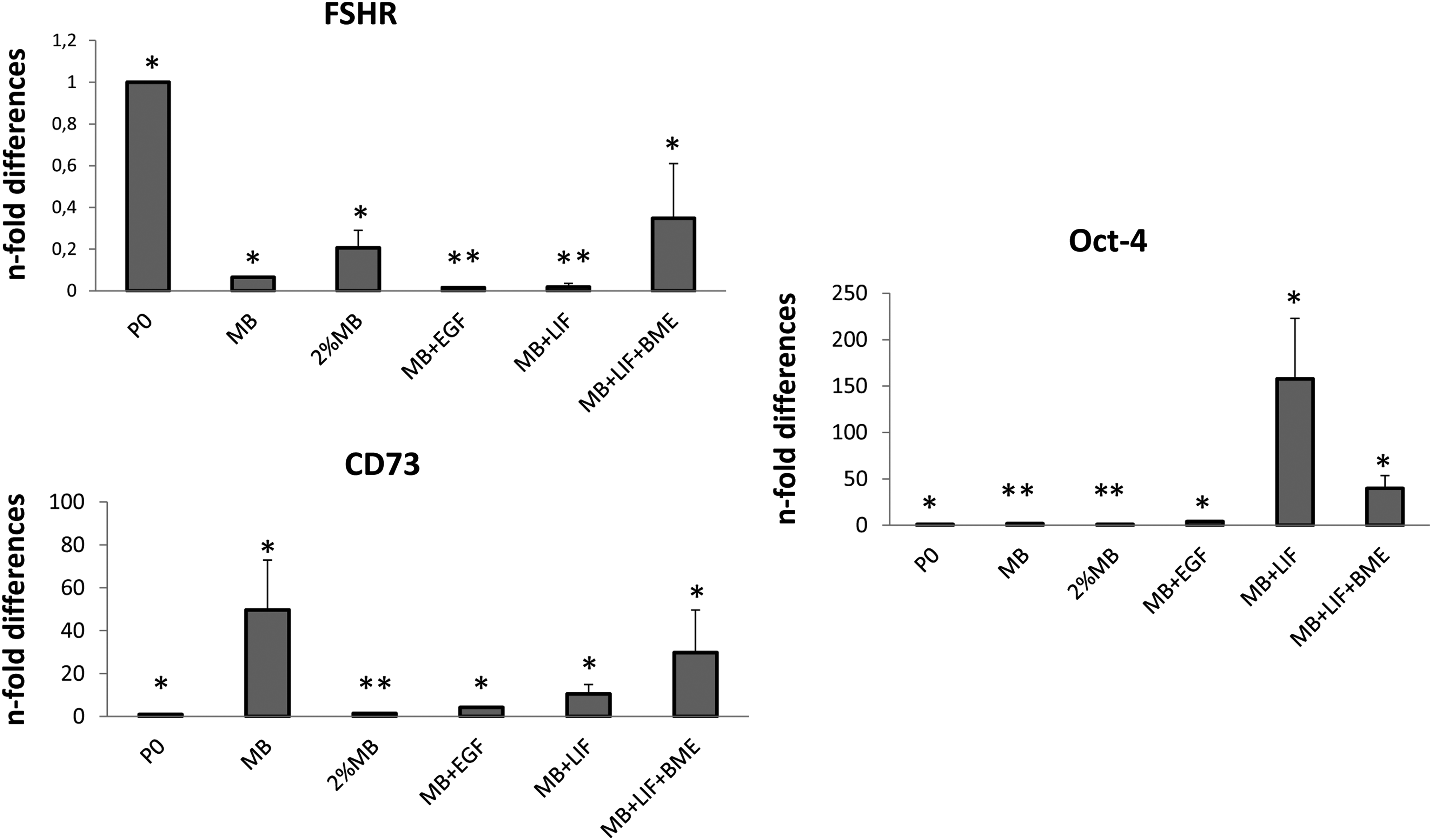
Quantitative RT-PCR analysis for the expression of multipotent (CD73), pluripotent (Oct-4), and granulosa-specific (FSHR) markers in cells cultured under different conditions (MB, 2% MB, MB + EGF, MB + LIF, MB + LIF + BME) at P3. Expression levels were normalized to the reference gene (GAPDH). Data are represented as fold change compared with expression observed in P0. Values are mean ± standard deviation (SD) (n = 3). Asterisks depict highly significant (**p < 0.01) differences.
GCs before and after acidic treatment expressed Oct-4, C-Myc, CD73, CD29, CD44, MHC I, MHC II, and granulosa-associated markers (FST and LIFR), but not CD34. The main difference found between d0 and d7 was the loss of FSHR expression (Fig. 4A). Considerable differences in gene expression were observed in GCs before and after acidic treatment (Fig. 4B). In particular, FSHR expression was found significantly decreased in treated cells at d7. Further indications supporting the efficacy of the treatment were provided by the loss of other granulosa markers (i.e., FST and LIFR) and the increase of Oct-4 and CD29 compared to untreated cells (d0). Decreased expression of MHC I and MHC II in d7 was also observed. In addition, as a response to the acidic treatment, in bovine GCs expression of inflammatory markers, such as TNF-α and IL-1β, was found to be significantly upregulated at d7 compared to d0, with a 7.02- (± 0.2) and 21.26- (± 0.25) fold increase (Fig. 5; p < 0.05 and p < 0.001, respectively).
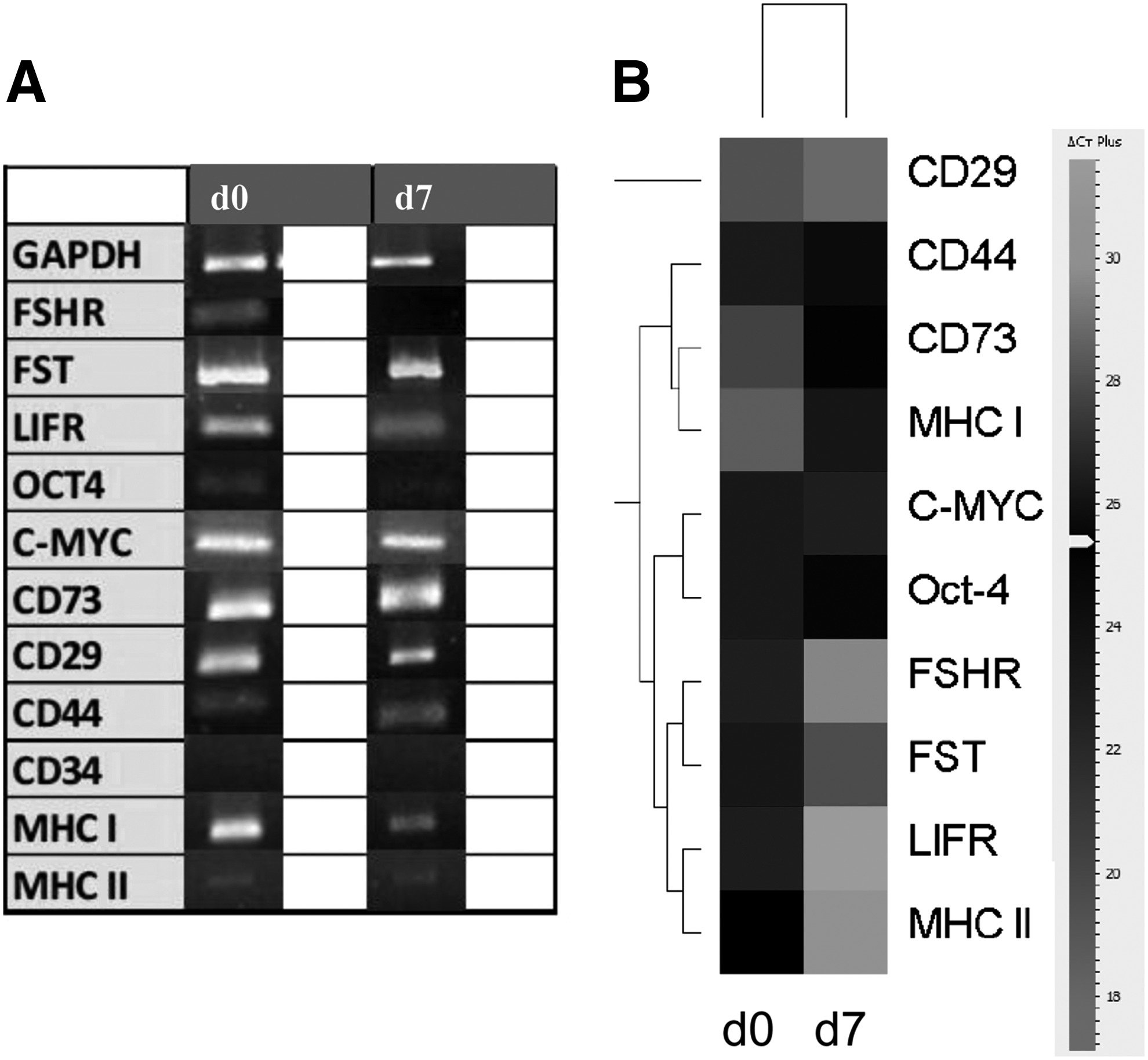
Qualitative (
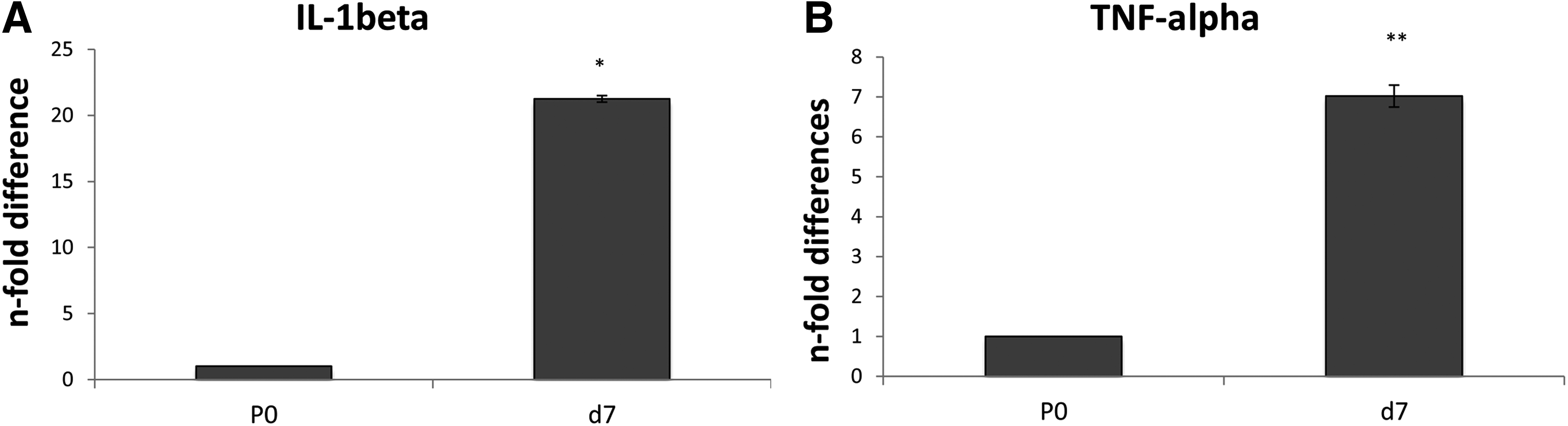
Quantitative RT-PCR analysis for the expression of inflammatory markers such as TNF-α and IL-1β in GCs before (d0) and after acidic treatment (d7). Expression levels were normalized to the reference gene (GAPDH). Values are mean ± standard deviation (SD) (n = 3). Asterisks depict highly significant (**p < 0.01) and significant (*p < 0.05) differences compared to d0.
In vitro differentiation
GCs cultured in different conditions did not show any ability to differentiate (data not shown). On the other hand, GCs treated with an acidic pH were easily induced into the adipogenic, osteogenic, and neurogenic lineages. After 18 days of induction, the presence of intracellular lipid vacuoles was determined by Oil Red O staining (Fig. 6A). After 21 days in osteogenic media, extracellular mineral deposits were demonstrated by von Kossa staining (Fig. 6B). Interestingly, cells induced to differentiate toward the neurogenic lineage acquired the typical neuronal morphology with axon- and dendrite-like processes and were positive for Nissl staining of Nissl bodies (Fig. 6C). Uninduced cells were maintained in culture for the same period of each differentiation protocol time and used as a negative control. They were negative for all of the staining performed. Differentiation was confirmed by molecular analysis through the use of specific markers, including leptin (LEP) and peroxisome proliferator-activated receptor gamma (PPARγ) for adipogenic differentiation, bone gamma-carboxyglutamate protein (BGLAP), osteopontin (SPP1), and secreted protein acidic and rich in cysteine (SPARC) for osteogenic differentiation, and glial fibrillary acidic protein (GFAP) for neurogenic differentiation.

Differentiative potential. (
In cells induced to undergo adipogenesis the expression of PPARγ but not LEP was revealed in induced cells, whereas control cells did not express the genes tested. Following osteogenesis induction, BGLAP was not expressed in induced cells. Qualitative PCR did not allow for the discrimination between induced and uninduced cells when the expression of SSP1 and SPARC was assessed (Fig. 6B). For this reason, qPCR was performed to determine the levels of expression of these two markers in induced cells compared to their respective uninduced controls. The expression of osteogenesis-associated genes quantitatively confirmed the induction. SPP1 expression increased 2.17- (±0.09) fold (p < 0.05), whereas a slight but statistically significant (p < 0.05) increase (1.45 ± 0.085) in SPARC expression was found compared to the uninduced counterparts (Fig. 7). The expression of GFAP confirmed the neurogenic differentiation.
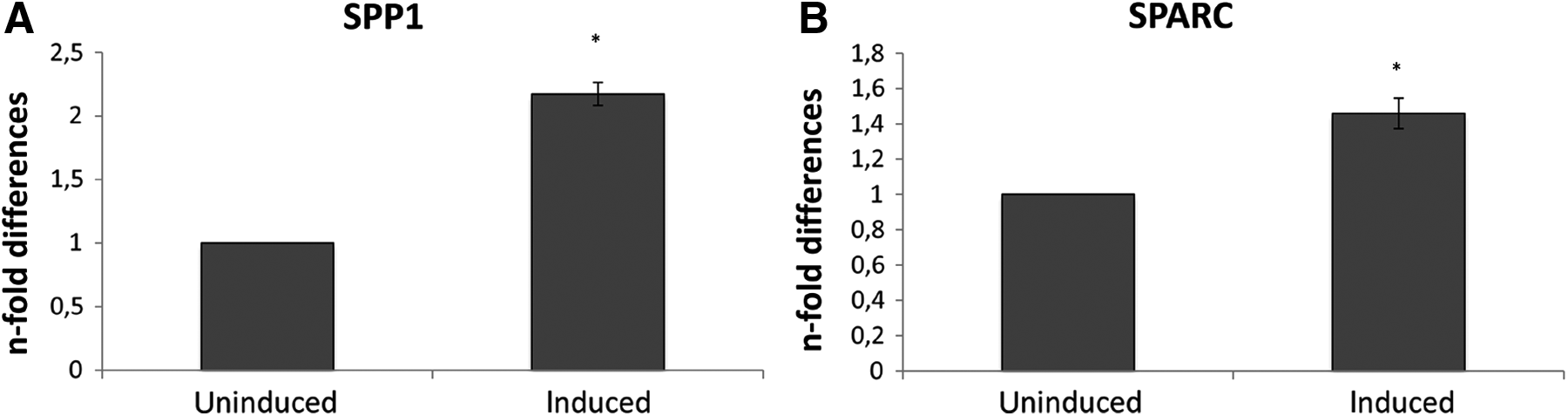
Quantitative RT–PCR analysis for the expression of osteogenesis-associated genes such as SPP1 (
Immunocytochemical characterization of Oct-4
Immunopositivity to Oct-4 was detected in GCs cultured in MB + LIF and in bovine embryos used as a positive control. GCs after collection, used as a control, were negative (Fig. 8).
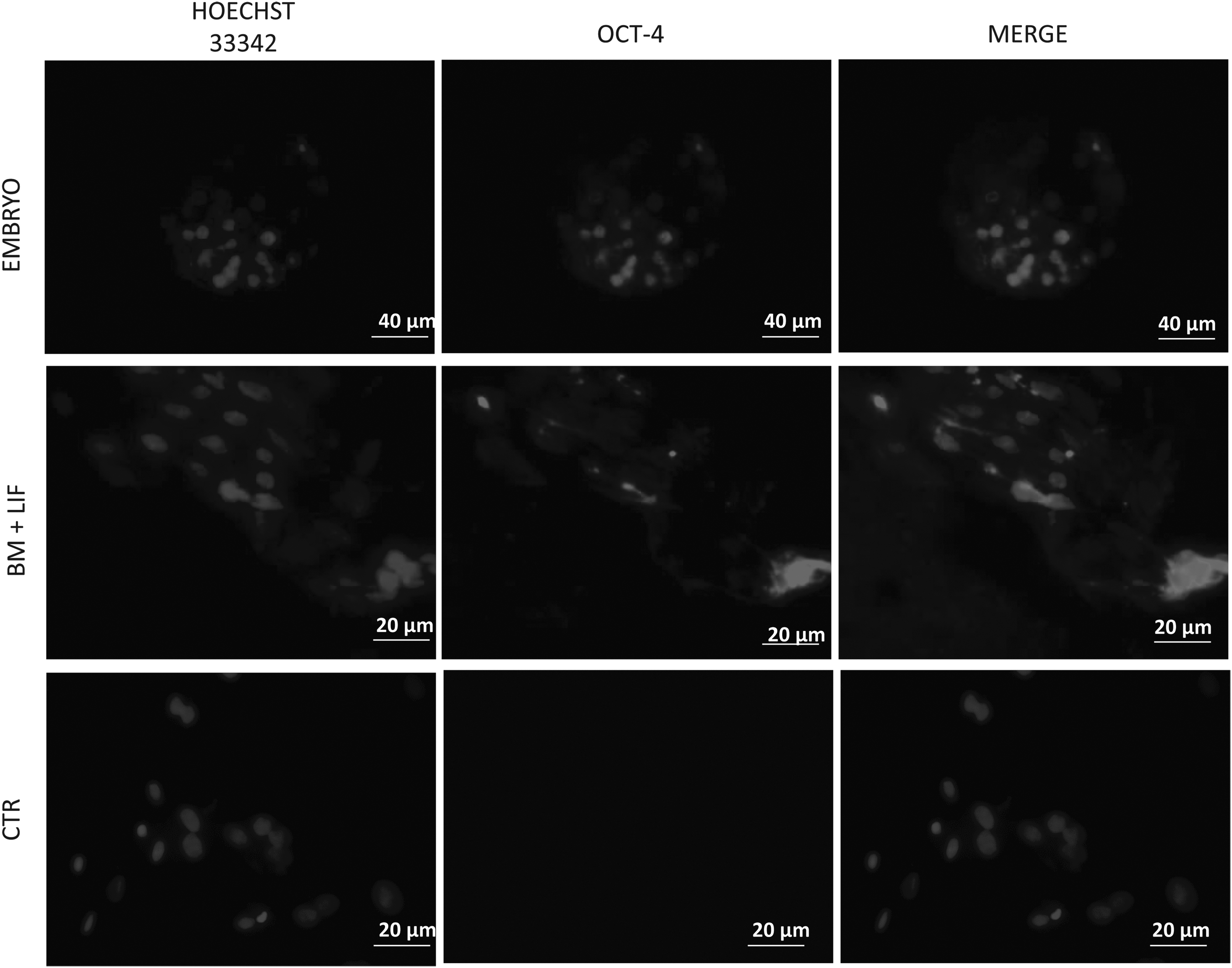
Photomicrographs of immunostaining of bovine GCs labeled with antibodies against antigens for Oct-4. Magnification, 40×; scale bar, 20 μm. Magnification for embryos: 20×; scale bar, 40 μm. CTR, control.
Discussion
The purpose of this study was to identify a new source of stem cells that were easy to collect and able to comply with the requirements of regenerative medicine on a large scale. On the basis of recently published studies in humans (Kossowska-Tomaszczuk et al., 2009) and gilts (Mattioli et al., 2012), GCs represent an alternative source of MSCs.
To reach our goal, we tested different culture conditions and evaluated their efficacy in selecting MSCs from a cell population obtained from the follicle. Once isolated, GCs have been characterized on the basis of the minimal criteria defined by the International Society for Stem Cell Therapy to define MSCs (Dominici et al., 2006). According to results obtained in humans (Kossowska-Tomaszczuk et al., 2009) and gilts (Mattioli et al., 2012), cells adhered to a plastic dish and expressed a pattern of mesenchymal (CD44, CD29, CD166, CD73) and pluripotency (Oct-4, c-Myc) genes, with no expression of the hematopoietic CD34 and the functional marker FSHR. The levels of expression of CD73, Oct-4, and FSHR were also confirmed quantitatively, showing the reduction of specific granulosa markers and the upregulation of MSC-associated genes.
In addition to molecular characterization by RT-PCR, the high level of Oct-4 in GCs cultured in MB + LIF was also detected by immunocytochemistry. Contrary to data reported for humans (Kossowska-Tomaszczuk et al., 2009) and gilts (Mattioli et al., 2012), however, despite inducing a MSC-like phenotype in bovine GCs, none of the culture conditions tested was efficient in selecting a plastic cell population. In fact, cells exposed to each condition were not able to differentiate into adipogenic, osteogenic, and neurogenic lineages. To date, the expression of FST, a protein broadly secreted by GCs in the ovary, was never found to be downregulated, as we would expect it to be for cells retaining/acquiring a stem cell–associated phenotype. The FST gene continued to be switched “on” in the genetic pattern of GCs, even if they were grown under different culture conditions. Its expression probably indicates an incomplete dedifferentiation to mesenchymal cells and the maintenance of a strong epigenetic imprinting that prevented GCs from differentiating. This epigenetic imprinting could be the reason of failed differentiation.
On the basis of our findings, it is reasonable to assume that GCs isolated in our study and maintained in different conditions can be considered progenitors of GCs; the only exception was those kept in MB + LIF + BME that lost expression of CD44 and CD166. The discrepancy in the differentiation outcome observed between the present study and that reported by Mattioli et al. (2012) and Kossowska-Tomaszczuk et al. (2009) is difficult to explain considering that GCs were isolated from the internal side of the preovulatory follicle and, thus, from the periantral layer, as reported by Mattioli et al. (2012). Erickson (2000) postulated that stem cells could be located in the periantral layer of granulosa. As such, this method could be the most appropriate for the collection of multipotent stem cells from granulosa compared to the aspiration protocol performed by Kossowska-Tomaszczuk et al. (2009).
On the basis of our negative results to detect stem cells in preovulatory follicles, we decided to expose GCs to chemical stress by lowering the pH in culture to elucidate whether acidic stress could influence the phenotype and, eventually, help with the selection of more plastic cells. In plants, drastic environmental changes have been reported to convert mature somatic cells (for example, dissociated carrot cells) into immature blastema cells, from which a whole plant structure, including stalks and roots, develops in the presence of auxins (Thorpe, 2007). In our study, molecular analysis revealed significant differences between the cells before and after the acidic treatment. In particular, 7d cells showed a significant decrease of expression of granulosa-specific markers (FSHR, FST, LIF-R), with a concomitant increase in the pluripotency-associated marker Oct-4. Chiou et al. (2008) also reported the upregulation of Oct-4 in these conditions. The loss of FSHR could be explained with the lack of its ligand (FSH) in the culture media, which is in agreement with the findings obtained by Kossowska-Tomaszczuk et al. (2009), focusing on human GCs isolated from the ovarian follicles of infertile patients and cultured in the presence of LIF.
Changes in gene expression can be further justified by hypothesizing that the chemical stress is able to induce either a cell dedifferentiation or a strong selection of progenitor cells. Changes in the external environment (including the pH reduction) have been previously associated with a specific phenotype acquired by cells exposed to them, as in the case of ovary cells and cancer cells (Tannock and Rotin, 1989). Moreover, when cultured in acidic conditions, cells lose the expression of the immunogenic markers (MHC I and MHC II), confirming their more undifferentiated state. MSCs have been reported to be immuno-privileged cells, with no or low expression of those markers (Hass et al., 2011).
Differentiation studies further corroborated such observations. pH-treated cells were induced toward the mesodermic (adipogenic and osteogenic) and ectodermic (neurogenic) lineages. Oil Red O, von Kossa, and Nissl staining, respectively, demonstrated differentiation, which was further confirmed by molecular analysis. For adipogenesis, we investigated the expression of PPARγand LEP; however, only the first one was expressed in differentiated cells. The expression of PPARγ suggests a preadipocytic commitment of cells (Corradetti et al., 2013), which is further confirmed by the lack of LEP, a marker regarded as an intermediate and late marker. The expression of GFAP in induced cells suggests that astrocyte differentiation occurred, as previously reported for bovine and equine amniotic-derived cells (Corradetti et al., 2011, 2013; Lange-Consiglio et al., 2012).
Osteogenesis was assessed investigating the osteogenic-specific markers BGLAP, SPP1, and SPARC. All of these markers were expressed in induced cells, with the exception of only BGLAP. This might be explained considering that BGLAP is expressed in terminally differentiated osteoblasts (Wagner et al., 2011). Surprisingly, the expression of SPP1 and SPARC was detected also in the negative controls (7d uninduced cells), although with a lower expression level compared to induced cells. We hypothesized that the expression of these markers (SPP1 and SPARC) in negative controls can be due to the role they play in inflammation. Indeed, these markers are mainly involved in the immune response to an inflamed environment (Lund et al., 2009; Xu et al., 2013), as is the case of the acidic treatment. The upregulation of TNF-α and IL-1β expression confirmed our hypothesis. In particular, SPARC levels are significantly correlated with inflammation (Xu et al., 2013) and SPP1 is strikingly upregulated at sites of inflammation and tissue remodeling, because it promotes the migration of inflammatory cells to the wound site and functions as a proinflammatory cytokine (Lee et al., 2012).
Conclusions
Results obtained from this work demonstrated that none of the culture conditions employed in this study allowed for the selection of the stem cell population within GCs isolated by scraping. The stress induced by the acidic treatment on bovine GCs endorsed the selection of the more plastic cells, which were the only ones able to respond to stimuli and adjust to a more rigid environment. Thus, compared to the freshly isolated cells, selected cells showed an increased expression of the pluripotent marker Oct-4 and were able to differentiate into mesodermic and ectodermic lineages. The acquired phenotype of those cells can be also explained as a consequence of the activation of an inflammatory process, able to determine de-differentiation or nuclear reprogramming in GCs (Lee et al., 2012). Further studies are required to understand better the effect of the acidic treatment and the consequent stress induced by it at molecular level. Moreover, different approaches will be required to discover a possible stem cell niche in bovine pre-ovulatory follicle.
Footnotes
Acknowledgments
This study was supported by grants from Università degli Studi di Milano and Università Politecnica delle Marche, Ancona, Italy. The authors wish to thank Prof. Fulvio Gandolfi and Dott.ssa Georgia Pennarossa (Department of Veterinary Science Animal Health and Food Safety, Univerità degli Studi di Milano, Italy) for providing the reagent and protocol for Oct-4 immunocytochemical detection.
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
Author contributions: Anna Lange-Consiglio is responsible for the study concept and participated in designing the study, performed the in vitro study, collected and interpreted data performing statistical analysis, wrote and reviewed the manuscript, and approved the final version; Alessio Romaldini performed the molecular study, collected and interpreted data, and approved the final version; Alessio Correani performed the molecular study, collected and interpreted data, and approved the final version; Bruna Corradetti performed the molecular study, collected and interpreted data, wrote and reviewed the manuscript, and approved the final version; Paola Esposti took part in collecting and interpreting data and approved the final version; Maria Francesca Cannatà took part in collecting and interpreting data and approved the final version; Claudia Perrini took part in collecting and interpreting data and approved the final version; Maria Giovanna Marini took part in collecting and interpreting data and approved the final version; Davide Bizzaro took part in collecting and interpreting data and approved the final version; Fausto Cremonesi is responsible for the study concept and participated in designing the study, interpreted data, and revised and approved the final version of the manuscript.