
Abstract
Abstract
Buffalo embryos were produced by hand-made cloning using skin fibroblasts from male and female buffaloes (n = 4 each) as donor cells for examining the effect of sex. Although the rate of blastocyst formation (43.8% ± 1.31% vs. 42.2% ± 1.22%) was similar, the total cell number (333 ± 10.4 vs. 270 ± 10.9) was higher (p < 0.05) whereas the apoptotic index (6.39 ± 0.25 vs. 8.52 ± 0.38) was lower (p < 0.05) for male than for female blastocysts. In the blastocysts, the global level of H3K18ac was found to be in the following order: male>female>IVF (in vitro fertilization) blastocysts (p < 0.05). The global level of H3K9me2 was not significantly different between male and female blastocysts and was higher (p < 0.05) compared with that in their IVF counterparts. The relative mRNA abundance of X-chromosome-linked (XIST, HPRT, PGK, and G6PD), apoptosis- (CASPASE3) and pregnancy-related genes (IFN-τ) was significantly higher (p < 0.05) whereas that of DNMT1 was significantly lower (p < 0.05) in female than in male blastocysts; however, in the case of apoptosis- (BCL-XL) and developmental competence-related genes (IGF1R and OCT4), the expression level was similar between the two groups. The gene expression level of OCT4 and IFN-τ but not of IGF1R was significantly lower (p < 0.05) in cloned than in IVF blastocysts. This study demonstrates that the epigenetic status, quality, and expression level of several genes but not the developmental competence are affected by the sex of cloned embryos.
Introduction
E
Aberrant epigenetic reprogramming is considered the primary reason that is responsible for the low success rate of cloning (Niemann et al., 2008). The epigenetic defects in cloned embryos can be broadly classified into the following three categories: (1) hypermethylation of genes; (2) histone acetylation and methylation, which involve the whole set of genes present on autosomes and sex chromosomes; and (3) errors in X-chromosome inactivation (XCI), which affect the sex chromosome of only females (Kang et al., 2001). XCI, which is an epigenetic hallmark of mammalian development, is a mechanism for dosage compensation that operates in the early phases of embryonic development and leads to one of the X-chromosomes being silenced in female mammals. There are two copies of genes located on X-chromosome in females compared with only one copy present in males. The occurrence of XCI in female embryos makes sure that the male (XY) and female (XX) embryos contain equal dosages of X-chromosome-linked genes during female embryogenesis (Lyon, 1961).
Aberrations in XCI have been observed in NT embryos and offspring. Abnormalities that have been reported to be present at various developmental stages include both the X-chromosomes being active in some cells of cloned blastocysts, XCI being absent in late-stage placenta, and so on (Nolen et al., 2005). Aborted bovine NT fetuses as well as dead newborn calves showed aberrant patterns of XCI (Xue et al., 2002). Absence of occurrence of XCI in extraembryonic tissues resulted in causing long-term growth impairment and decreased survival in clones (Eggan et al., 2000; Xue et al., 2002).
Very few reports are available in which the developmental competence, epigenetic modifications, and level of expression of some important genes have been compared between male and female NT blastocysts (Amarnath et al., 2007). This study aimed at comparing the developmental competence and embryo quality of male versus female NT buffalo embryos. The rate of blastocyst formation was taken as a measure of the developmental potential, whereas the total cell number (TCN) and the apoptosis level measured by TUNEL assay were used as parameters for determining the quality of embryos. Epigenetic status was also compared between male and female blastocysts in terms of the global levels of histone modifications H3K18ac and H3K9me2. In addition, the relative mRNA abundance of some X-chromosome- (XIST, G6PD, HPRT, and PGK), apoptosis- (BCL-XL and CASPASE3), development- (IGF1R and IFN-τ), pluripotency- (OCT4), and epigenetic status-related genes (DNMT1) was also compared between male and female NT blastocysts by using their IVF (in vitro fertilization) counterparts as controls.
Materials and Methods
All the chemicals and media used in the present study were obtained from Sigma Chemical Co. (St. Louis, MO); whereas all the plasticware was purchased from Nunc (Rosklide, Denmark), unless indicated otherwise. Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT). Approval of the Animal Ethics Committee of NDRI, Karnal, was obtained before carrying out animal experiments. Four adult male (Mu-5710, Mu-5926, Mu-6044, and Mu-6136) and an equal number of adult female Murrah buffaloes (Mu-5579, Mu-5924, Mu-5517, and Mu-5365) available at Livestock Research Centre of NDRI, Karnal, were used for obtaining donor cells.
Establishment and characterization of somatic cells
Skin biopsies were obtained by punching the ear pinna of experimental animals, after which the cells were established in culture as already described by Selokar et al. (2012a). The cells were sub-cultured up to 10 times. Cells at early passages (P2–3) were stored frozen in liquid nitrogen in freezing medium that consisted of DMEM/F12 + 10% dimethyl sulfoxide +20% FBS for use in future.
Expression of various cytoskeleton markers was examined by immunofluorescence staining as described by Selokar et al. (2014), with some modifications. Briefly, early passage (2–3) cells were seeded in 96-well cell culture plates at a density of ∼2000 cells/well and were cultured till 70%–80% confluence was achieved. The cells were treated with 4% paraformaldehyde for 1 hour for fixing, then with 0.5% TritonX-100 for 30 minutes for permeabilization, and finally with 3% bovine serum albumin (BSA) for blocking. This was followed by incubation for 1 hour with the primary antibody (mouse anti-cytokeratin, 1:50, MAB1611; Millipore, Billerica, MA; mouse anti-vimentin 1:500, V6630; Sigma) that was diluted by using the blocking solution. The cells were then incubated for 1 hour with the secondary antibody (goat anti-mouse/rabbit IgG) that was conjugated with fluorescein isothiocyanate (FITC). Separate controls (negative and positive) were run in each respective cell culture for testing the staining protocol. The positive controls were labeled with mouse anti-tubulin (1:500, T8328; Sigma), whereas the primary antibody was omitted in case of the negative controls. The cells were treated with 10 μg/mL propidium iodide (PI) for 5 minutes for staining the nuclei. Fluorescence in the cells was detected by epifluorescence microscopy. Adobe Photoshop CS 8.0.1 software (Adobe Systems, Inc., San Jose, CA) was used for merging the images.
BrdU cell proliferation assay
The cell proliferation rate was determined by BrdU cell proliferation assay as described earlier (Mohapatra et al., 2015b). This assay is based on the fact that actively proliferating cells incorporate 5-bromo-2′-deoxyuridine-5′-triphosphate (BrdU, B5002-250 MG; Sigma) into their DNA. Briefly, the somatic cells were seeded in a 96-well plate, after which 10 mMBrdU label (10 μL/well) was added and the plate was incubated in a CO2 incubator at 37°C for 24 hours. Then, for fixing the cells, chilled methanol was added and the cells were incubated for 20 minutes at −20°C. The cells were then incubated with 1% TritonX-100 for 1 hour for permeabilization, and finally with 5% BSA for 1 hour for blocking.
This was followed by addition of monoclonal anti-BrdU raised in mouse (B8434-200UL; Sigma) to all the wells except the negative control wells, after which the plate was incubated overnight at 4°C. Each well was then washed thrice with 0.1% Triton X-100 in DPBS (DPBST), after which the plate was incubated for 1 hour with FITC-conjugated anti-mouse IgG. After washing the wells thrice with DPBST, each well was supplemented with the nuclear stain (PI or Hoechst 33342) and antifade solution. The cells were examined for fluorescence under a fluorescence microscope. The proliferation index was calculated by using the following equation: the number of BrdU-positive nuclei in a given field/total number of nuclei counted in that field × 100. All the experiments were repeated at least thrice and had two replicates each.
Production of embryos by hand-made cloning (HMC)
Immature buffalo oocytes were collected from slaughterhouse ovaries, subjected to in vitro maturation, after which HMC was performed as described earlier by Selokar et al. (2012b) with some changes. In brief, after in vitro maturation, hyaluronidase treatment was given by using 0.5 mg/mL hyaluronidase in T2 medium (in which T stands for TCM-199 supplemented with 2.0 mML-glutamine +0.2 mM sodium pyruvate +50 μg/mL gentamicin, and 2 stands for 2% FBS) for removal of cumulus cells from cumulus-oocyte complexes. The cumulus-free oocytes were then incubated for 10 minutes with pronase (2.0 mg/mL in T20 medium) to remove the zona pellucida.
For visualization of the protrusion cone, the oocytes were incubated for 15–20 minutes in a CO2 incubator at 38.5°C in T20 medium. The oocytes were then bisected manually with a microblade after being transferred to cytochalasin-B (2.5 μg/mL in T20) for removal of the nuclear material. The larger demicytoplasts were incubated with phytohemagglutinin (0.5 mg/mL in T2) for 3–4 seconds and were immediately transferred to T2, which contained the donor cells dispersed at a low cell density. Each demicytoplast was then attached to a single donor cell by rolling. The donor cell–demicytoplast pairs were incubated for 5 minutes in the fusion medium, which consisted of 0.3 M D-mannitol +0.05 mM CaCl2 + 0.1 mM MgCl2 + 1 mg/mL polyvinyl alcohol (PVA), after which an A.C. pulse of 4 V was given through a BTX Electrocell Manipulator 200 (BTX, San Diego, CA) in such a way that the negative electrode was faced by a donor cell. Another demicytoplast was then immediately placed close to the donor cell in the fusion chamber (BTX microslide 0.5 mm gap, model 450; BTX). The triplets were fused by applying a single D.C. pulse of 3.36 kV/cm for 4 seconds, after which these were incubated for 6 hours in T20. For activation, the reconstructed embryos were incubated with 4 μM calcimycin A23187 in T20 for a duration of 5 minutes at 38.5°C, after which these were washed three times with T20.
Then, each reconstruct was shifted to droplets (5 μL each) of 2 mM 6-dimethylamino purine in T20, overlaid with mineral oil, and incubated for 4 hours in a CO2 incubator. Groups of 15–20 embryos were transferred to each well of a four-well dish containing 400 μL of the in vitro culture (IVC) medium (RVCL medium +1% fatty acid-free BSA), overlaid with mineral oil, and incubated for a period of 8 days in a CO2 incubator at 38.5°C.
Production of embryos by in vitro fertilization (IVF)
Immature buffalo oocytes were aspirated from buffalo ovaries that were procured from a nearby slaughterhouse. In vitro maturation (IVM) and IVF were carried out as previously described (Sharma et al., 2011). IVC of presumptive zygotes was carried out for the first 2 days postinsemination (dpi) in Charles Rosenkrans medium containing amino acids (mCR2aa) that was supplemented with 0.6% BSA and, subsequently, in 100 μL droplets of IVC medium (mCR2aa +10% FBS +0.6% BSA) on the original monolayer of granulosa cells for up to 8 dpi at 38.5°C in a CO2 incubator. Every 48 hours, 50% of medium was replaced with fresh medium. The blastocyst formation rate was measured 8dpi.
TUNEL assay
For assessing the quality of day 8 blastocysts, apoptosis level was measured by TUNEL assay using an In Situ Cell Death Detection Kit, Fluorescein (11684795910; Roche) following the protocol described by Mohapatra et al. (2015b), with minor modifications. The blastocysts were washed thrice with PVA (0.3% in PBS), after which these were treated for 1 hour with paraformaldehyde (4%) for fixing, and afterward with Triton X-100 (0.5% in PBS) for 1 hour for permeabilization. The blastocysts were then incubated with FITC-conjugated dUTP and terminal deoxynucleotidyltransferase (TdT) enzyme for1 hour at 37°C in the dark and then subjected to treatment with RNase (50 μg/mL), after which these were stained with 10 μg/mL nuclear stain Hoechst 33342 for 20 minutes at 37°C.
Blastocysts taken as positive controls were treated with DNase solution (100 U/mL) for 20 minutes at 37°C before incubating them with FITC-conjugated dUTP and TdT for 1 hour. The blastocysts were given several washings with Ca2+ and Mg2+-free DPBS before mounting them on glass slides in about 3 μL of antifade solution and flattening them by using a coverslip. The digital images obtained on an inverted fluorescence microscope were used for counting the cells. All the experiments were repeated at least three times. The apoptotic index of each embryo was calculated by using the equation: Apoptotic index in the blastocyst = (number of TUNEL-positive nuclei/total number of nuclei) × 100.
Immunofluorescence staining for epigenetic markers in cloned blastocysts
Immunofluorescence staining was used for examining H3K18ac and H3K9me2 global levels in HMC blastocysts. For this, the blastocysts were treated with paraformaldehyde (4%) at 37°C for 1 hour for fixing and afterward washed thrice with PVA (0.3% in Ca2+ and Mg2+-free DPBS). Permeabilization was carried out by treating the blastocysts with Triton X-100 (0.5% in Ca2+ and Mg2+-free DPBS) for 20 minutes at 37°C, after which blocking was done by treating them with BSA (3%) for 1 hour. Then, the blastocysts were given overnight incubation at 4°C with the respective primary antibody raised in rabbit (anti-H3K18ac, 1:1500 and anti-H3K9me2, 1:150; Millipore) diluted in BSA (3%).
After being washed five times with DPBST, the blastocysts were incubated with FITC-conjugated goat anti-rabbit secondary antibody (1:700; Sigma) and then, they were again washed five times with DPBST. The blastocysts were then incubated with H33342 (10 μg/mL) to counterstain the nuclei. After rinsing them with DPBST, the blastocysts were mounted on glass slides in DABCO (2.5% in glycerol) and then examined by using a fluorescence microscope. The optical conditions were kept the same for capturing the images. For acquisition of images and conversion of the mean pixel intensity emitted by each individual nucleus to a value, we employed the NIS-element basic research image processing software that the microscope (Nikon, Tokyo, Japan) was equipped with. For examining each epigenetic marker, we analyzed at least 10 images, with 200 nuclei from each image.
Quantitative real-time PCR
The relative expression level of various genes was examined by quantitative real-time polymerase chain reaction (qPCR) as previously described (Mohapatra et al., 2015a). In brief, we isolated total RNA from blastocysts (n = 10 each) by RNAqueous micro kit (Ambion, Austin, TX) according to the manufacturer's protocol. cDNA was prepared by carrying out an RT reaction by using Superscript Reverse Transcriptase III (Invitrogen) kit after DNase treatment. mRNA was quantified by qPCR using CFX 96 I Cycler (BioRad). The reaction mixture (10 μL) contained 10 μM of each primer (0.2 μL), SYBR Green master-mix (5 μL), and 2 × diluted cDNA. Thermal cycling conditions were as follows: initial denaturation at 95°C for 5 minutes, 40 cycles of 15 seconds at 95°C, 15 seconds at the corresponding annealing temperature, and 15 seconds at 72°C followed by 95°C for 10 seconds. PCR efficiency for all the primer pairs used was confirmed, and specific products were checked by melt curve analysis.
Appropriateness of size was checked by 2% agarose gel electrophoresis. The primer sequences have been presented in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/cell). For normalization of the expression data, GAPDH was taken as the housekeeping gene, which was analyzed with CFX Manager Software (BioRad). Three trials, each with three replicates, were carried out in all the experiments.
Experimental design and statistical analysis
Ear skin-derived cells from adult male and female buffaloes (n = 4 each) were subjected to isolation, establishment in culture, and characterization. First, we compared the proliferation rate of cells obtained from individual animals by using BrdU assay (Experiment 1). Then, we produced HMC embryos by using the cells obtained from individual male and female animals as donor cells (Experiment 2). We determined the rate of blastocyst formation as a measure of the developmental competence, whereas the apoptotic index and TCN, determined by TUNEL assay, were taken as a measure of the blastocyst quality. Then, we compared the global level of H3K18ac and H3K9me2 between the blastocysts produced from male and female animals and those produced by IVF (Experiment 3).
Finally, we compared the gene expression level of some X-chromosome- (XIST, G6PD, HPRT, and PGK), apoptosis- (BCL-XL and CASPASE3), development- (IGF1R and IFN-tau), epigenetics- (DNMT1), and pluripotency-related genes (OCT4) between male and female HMC blastocysts by using IVF blastocysts as controls (Experiment 4).
Sigma Stat version 3.1 (Aspire Software International, VA) was used for carrying out statistical analysis. Student's t-test or analysis of variance followed by Holm–Sidak test were used for analyzing datasets. Before analysis, percentage values were transformed according to arcsine transformation formula. Data were presented as mean ± SEM, and differences were considered statistically significant at p < 0.05.
Results
Ear skin-derived cells obtained from adult male and female animals (n = 4 each) were used for isolation of cells and establishment of cultures. Immunofluorescence staining was used to examine the expression of cytoskeletal markers for characterization of cells established from individual animals. It revealed their fibroblast nature, as the cells exhibited expression of vimentin but not cytokeratin (Fig. 1).
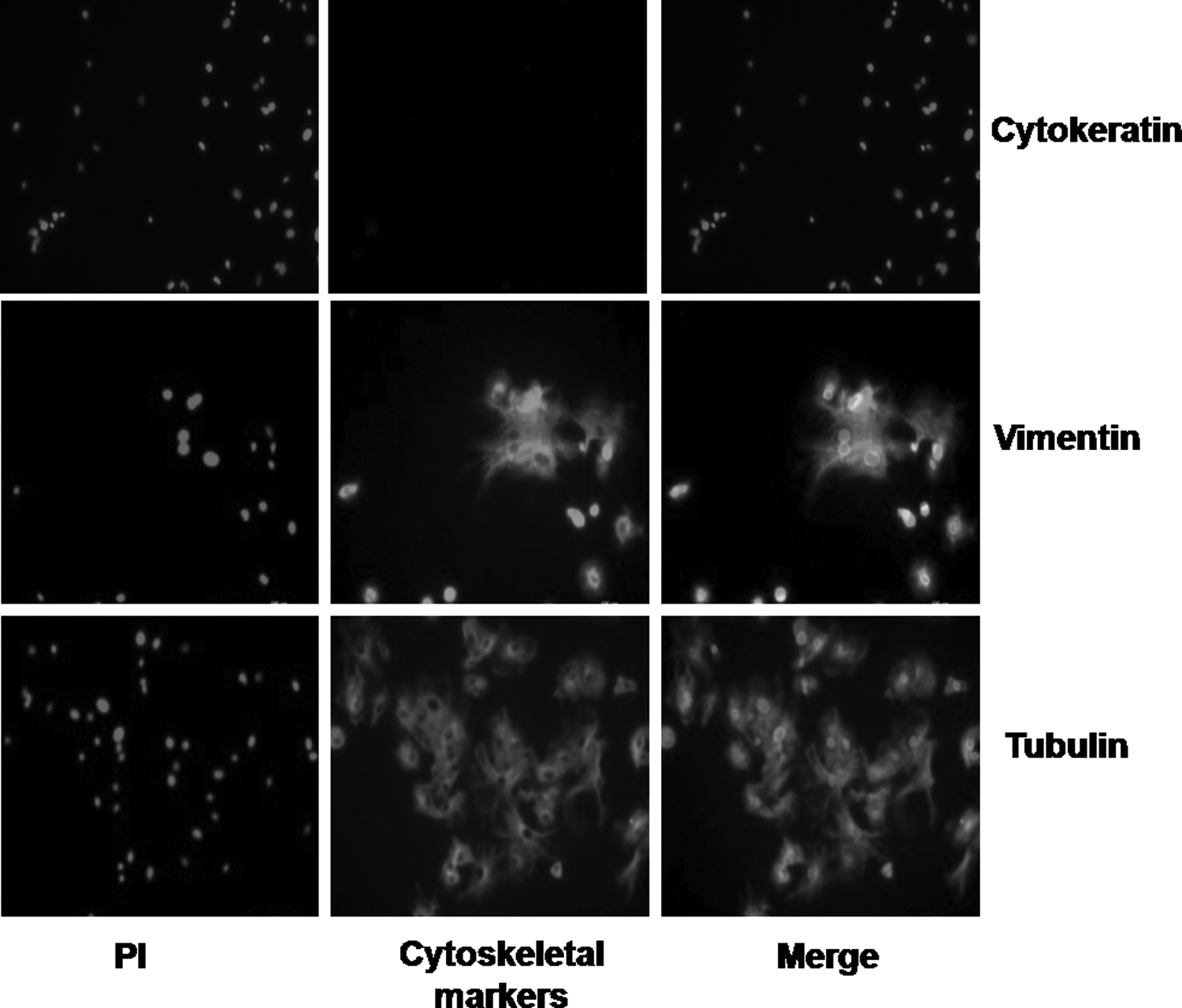
Donor cells showing expression of vimentin and tubulin but not that of cytokeratin after immunofluorescence staining, indicating that they were of fibroblast origin. PI, propidium iodide.
Experiment 1: proliferation rate of cell lines
A comparison of the proliferation rate of cell lines established from individual animals by BrdU assay revealed that, among male animals, the proliferation rate of cells obtained from Mu-6136 and Mu-5710 was higher (p < 0.05) than the proliferation rate of cell lines established from Mu-6044 and Mu-5926; however, among the female animals, the proliferation rate of cells differed significantly (p < 0.05) among the four individual animals (Fig. 2).

Proliferation rate of donor cells obtained from individual animals, measured by BrdU assay. Bars with different superscript letters differ significantly (p < 0.05).
Experiment 2: effect of embryo sex on quality and developmental competence
There were no significant differences in the cleavage (94.0% ± 0.50% and 92.9% ± 0.54%, respectively) and blastocyst rate (43.8% ± 1.31% and 42.2% ± 1.22%, respectively) between male and female embryos (Table 1). However, in terms of TCN, the blastocysts differed significantly (p < 0.05) in the following order: male HMC blastocysts>female HMC blastocysts>IVF blastocysts (333 ± 10.4, 270 ± 10.9, and 191 ± 13.3, respectively). The apoptotic index of male HMC blastocysts was lower (6.39 ±0.25) than that of female HMC blastocysts (8.52 ± 0.38); whereas that of IVF blastocysts (4.28 ± 0.53) was lower (p < 0.05) than the apoptotic index of male and female HMC blastocysts.
Data from 48 trials. Values are mean ± SEM. Values with different superscript letters within the same column differ significantly (p < 0.05).
IVF (in vitro fertilization).
Experiment 3: effect of embryo sex on epigenetic status
H3K18ac global level was significantly different (p < 0.05) in the following order: male HMC blastocysts>female HMC blastocysts>IVF blastocysts (Fig. 3). H3K9me2 global level, which was similar between male and female HMC blastocysts, was higher (p < 0.05) compared with that in their IVF counterparts.
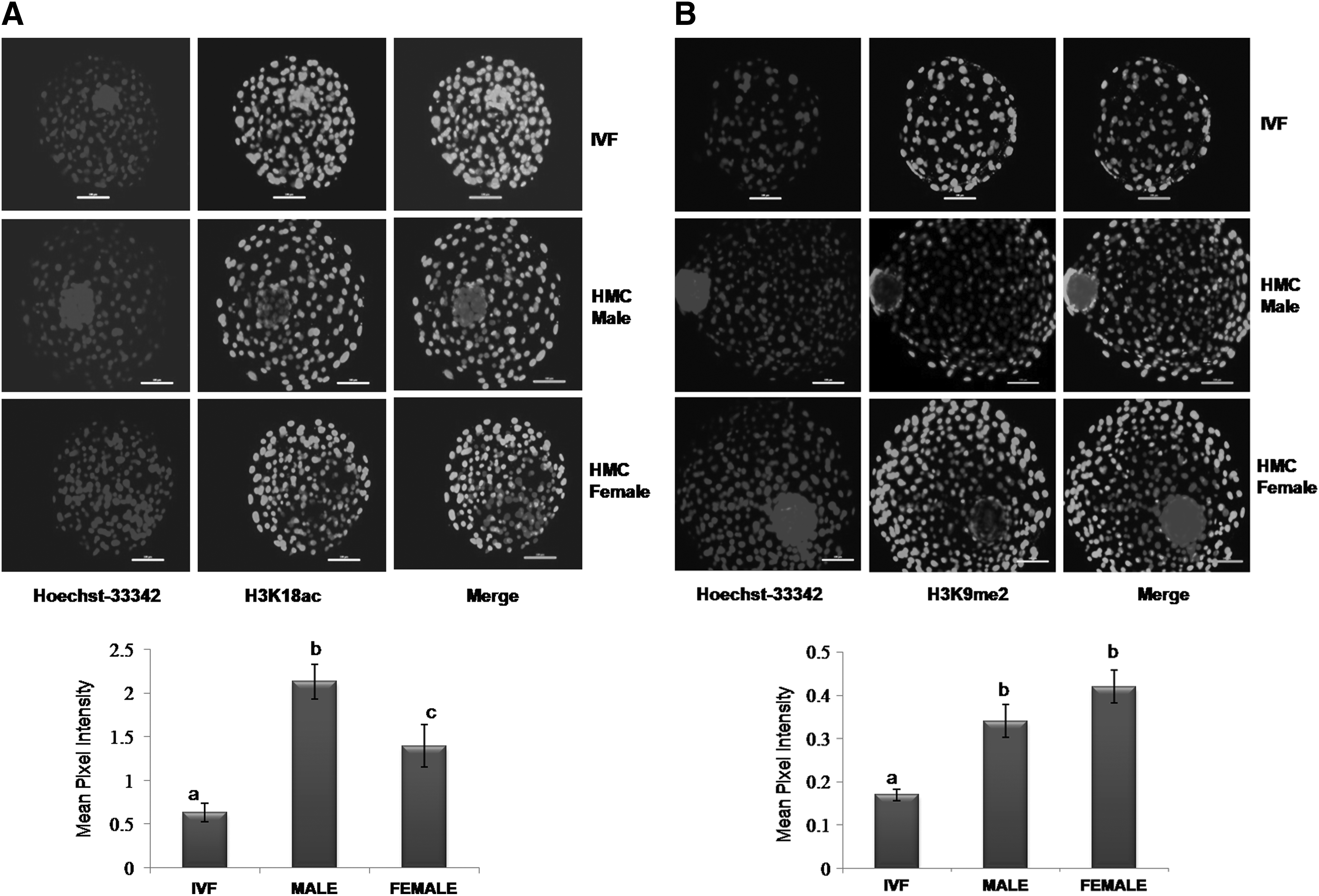
Mean pixel intensity of
Experiment 4: effect of embryo sex on gene expression
HMC female blastocysts exhibited a significantly higher (p < 0.05) relative expression level of XIST, an X-chromosome-linked gene, as compared with IVF blastocysts; however, its expression was absent in male HMC blastocysts (Fig. 4A). The expression level of other X-chromosome-linked genes HPRT, PGK, and G6PD was also significantly higher (p < 0.05) in female than in male HMC blastocysts (Fig. 4B). The relative mRNA abundance of CASPASE3, an apoptosis-related gene, was higher (p < 0.05) in female HMC blastocysts than in male HMC blastocysts and IVF blastocysts and that of BCL-XL was similar in the three groups (Fig. 4C). In case of the development-related gene IFN-τ, the relative expression level was significantly different (p < 0.05) in the following order: IVF blastocysts>female HMC blastocysts>male HMC blastocysts; however, that of IGF1R was similar in the three groups. The relative mRNA abundance of the pluripotency-related gene OCT4 was significantly higher (p < 0.05) in IVF than in male and female HMC blastocysts; whereas the expression level of DNMT1, an epigenetics-related gene, was significantly lower in female HMC blastocysts than in IVF and male HMC blastocysts in which it was similar.
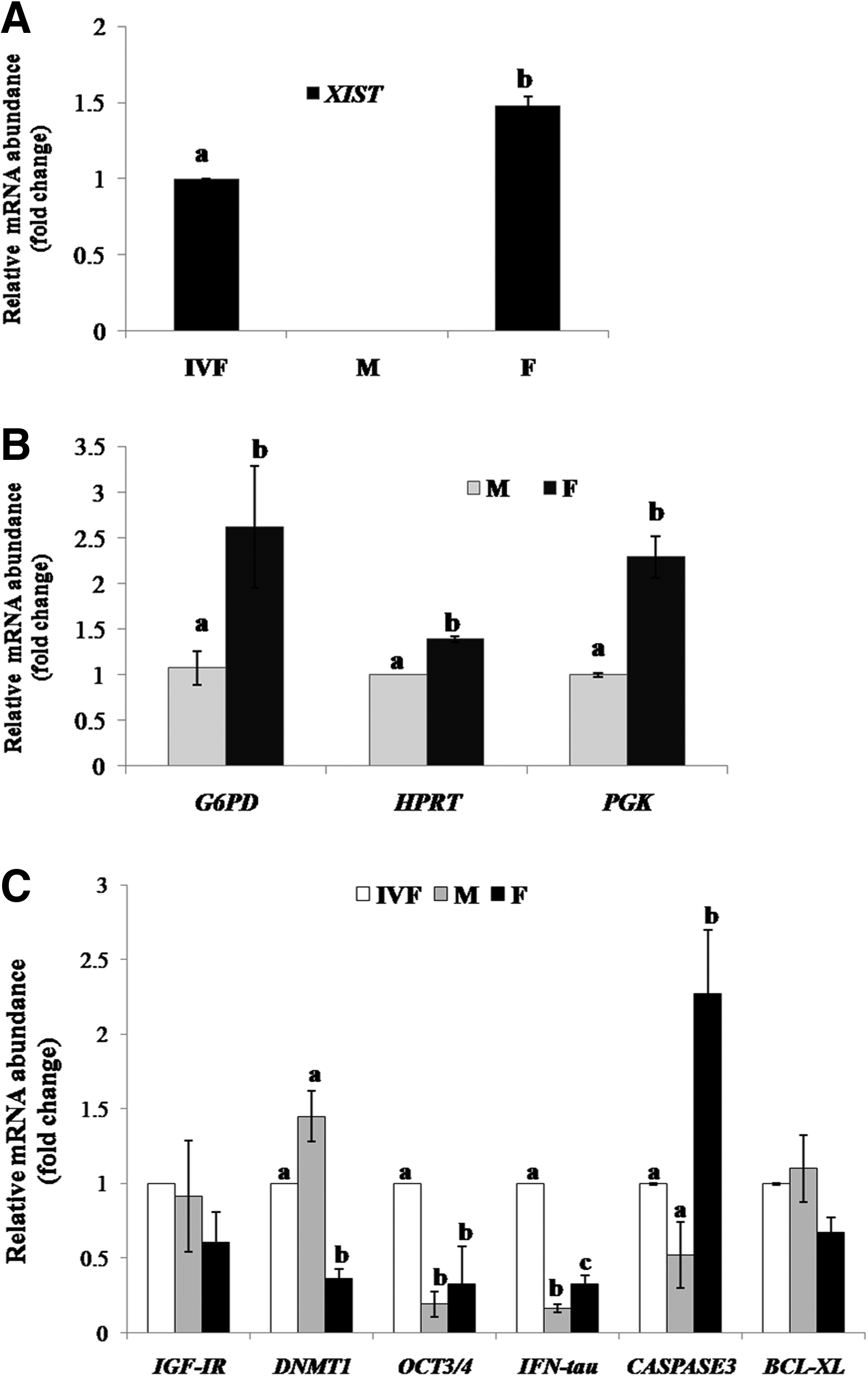
Relative mRNA abundance of
Discussion
We compared the developmental competence, epigenetic modifications, and relative mRNA abundance between male and female NT embryos produced by using the same type of donor cells, that is, skin fibroblast cells, to ensure that the differences in the epigenetic makeup of the donor cells did not interfere in the investigation. We found that despite similar developmental competence, male and female NT embryos differed in their TCN and apoptosis level, which are indicative of their quality. Male and female NT embryos also differed in terms of H3K18ac global level and the expression level of several genes.
First, we isolated skin fibroblasts from adult male and female buffaloes (n = 4 each), established them in culture, and examined the proliferation rate of cell lines established from individual animals by BrdU assay. Significant animal-to-animal variation was observed in the proliferation rate of cells obtained from both male and female animals, which agrees with our previous study (Selokar et al., 2014). Then, we produced HMC embryos by using the donor cells isolated from individual male and female animals. We found that the developmental competence, as indicated by the percentage of embryos cleaved, and the blastocyst formation rate did not differ between male and female NT embryos.
Similar results have been reported by Amarnath et al. (2007), who observed similar developmental rates to 2-, 4-, 8-cell, and blastocyst stage for bovine cloned embryos produced by using adult male skin fibroblasts and adult female cumulus cells as donor cells. This was also reflected in the expression levels of development-related gene IGF1R, which was similar among male and female NT blastocysts and IVF blastocysts, as also reported earlier by Amarnath et al. (2007). Our study further adds to these results by demonstrating that despite similar developmental competence, the quality of male embryos was superior to that of their female counterparts in terms of lower level of apoptosis and higher TCN. This is further substantiated by a significantly higher relative mRNA abundance of pro-apoptotic gene CASPASE3 in female as compared with male NT blastocysts in which it was similar to that in IVF blastocysts.
However, the expression level of the anti-apoptotic gene BCL-XL was similar among the three types of blastocysts. One of the important reasons for the lower conception rate obtained with NT compared with that with IVF embryos is believed to be the higher level of apoptosis in the former (Cui et al., 2011). Therefore, the lower apoptotic index of male NT embryos could be particularly important for in vivo development. However, further studies need to be carried out to find out whether the conception or offspring rate obtained with male and female NT embryos differs.
Reprogramming and the development of NT embryos are profoundly influenced by methylation and acetylation of histones, which are considered very important types of epigenetic modifications (Yamanaka et al., 2009). Therefore, we compared H3K18ac and H3K9me2 global levels between male and female NT blastocysts by using their IVF counterparts as controls. We found that H3K18ac global level was significantly different in the following order: male HMC blastocysts>female HMC blastocysts>IVF blastocysts. An increase in the global acetylation level of histones results in alleviation of transcription repression and, thus, facilitates remodeling of chromatin and relieves methylated CpG; whereas histone hyperacetylation makes nucleosomes more accessible to various factors (Jones et al., 1998; Lee et al., 1993). In contrast, methylation of H3 histone at K9 residue results in formation of constitutive heterochromatin, which is inactive (Rice and Allis, 2001).
It was found in our study that the global level of H3K9me2 was lower in IVF blastocysts than in male and female HMC blastocysts, which had a similar level. Although the specific reasons behind this are not clear, it signifies aberration in epigenetic modifications in NT embryos as compared with their IVF counterparts. In IVF embryos, methylation of H3K9 is associated with methylation of DNA. In a similar manner, hypermethylation of H3K9 is associated with hypermethylation of DNA, suggesting a genome-wide reprogramming failure in cloned embryos (Santos et al., 2003). After implantation, dimethylation of H3K9 plays an essential role in Oct4 inactivation through a novel multi-step program, indicating that H3K9 hypermethylation effects may not be confined to early embryonic development and may extend beyond that (Feldman et al., 2006). In live cloned cattle, distinct aberrance of H3K9me2 and H3K27me3 has been observed on the inactive X-chromosome (Geng-Sheng et al., 2009).
Transcriptional profiling of bovine blastocysts has shown that a majority of X-linked genes exhibit sex-related transcriptional differences and are involved in regulating the expression of autosomal genes in preimplantation embryos (Bermejo-Alvarez et al., 2010a). Many studies indicate that after NT, aberrant reprogramming has a profound influence on further development of cloned embryos, because it results in defects in X-chromosome regulation. Therefore, we studied the expression level of some important X-linked genes in male and female cloned blastocysts by using their IVF counterparts as controls. Among these, the importance of XIST, a noncoding RNA that is responsible for inactivating one of the X-chromosomes in females, has been established in many studies.
Cloned embryos have been reported to have impaired Xist regulation, leading to an increased risk of formation of defective placenta and high neonatal mortality rate (Hemberger, 2002; Xue et al., 2002). In cloned mouse embryos, an active X-chromosome was shown to express Xist ectopically and deletion of Xist from the active X-chromosome was shown to result in normal global expression of genes and about eight- to nine-fold enhancement of cloning efficiency (Inoue et al., 2010). Knockdown of Xist through RNAi has been shown to enable cloned mouse embryos to develop normally postimplantation, which was otherwise found to be impaired (Matoba et al., 2011). In our study, it was found that the relative expression level of XIST was higher in female NT as compared with IVF blastocysts whereas it was not detected in male NT blastocysts. Besides XIST, we studied the relative mRNA abundance of some other X-linked genes such as G6PD (glucose-6-phosphate dehydrogenase), PGK (phosphoglycerate kinase), and HPRT (hypoxanthine-guanine phosphoribosyltransferase). HPRT and G6PD are associated with metabolic pathways and reactive oxygen species detoxification (Peippo et al., 2002), whereas PGK is an enzyme that generates adenosine triphosphate during glycolysis (Latham et al., 2000).
It was found that the expression level of all of them was upregulated in female than in male NT blastocysts, which is in agreement with a recent report on pigs (Park et al., 2012). This indicates occurrence of partial XCI and absence of dosage compensation for expression of these genes in female NT embryos. The sex ratio is deviated more toward males in IVF embryos from the expected ratio of 50% in the embryos produced in vivo (Gutierrez-Adan et al., 1996; Wrenzycki et al., 2002). Bermejo-Alvarez et al. (2010b) reported that the expression level of most of the X-linked genes was higher in female than male bovine embryos, which is suggestive of partial XCI.
In contrast, the expression level of G6PD and PGK was similar between female and male bovine embryos produced in vivo (Wrenzycki et al., 2002). Specifically, the expression level of G6PD, the first and the most important pentose phosphate pathway regulatory enzyme, which may be responsible for differences in metabolism between male and female embryos, is higher in female than male IVF embryos (Gutierrez-Adan et al., 2000; Peippo et al., 2002; Wrenzycki et al., 2002). This may be the reason behind the deviated sex ratio observed in IVF embryos.
Since a high level of DNA methylation can result in reversible repression of genes, it is considered one of the most important epigenetic alterations. DNA methylation is carried out by DNA methyltransferases (DNMTs). Methylation of the newly formed DNA strand is carried out during S-phase of the cell cycle by DNMT1, which acts as a maintenance methyltransferase; whereas de novo methylation is established during gametogenesis and early stages of embryonic development, by DNMT3a and DNMT3b (Bestor, 2000). Our study revealed that the expression level of DNMT1 was higher in male than female NT blastocysts.
IFN-τ, the peripheral concentrations of which increase significantly at the time of implantation, is considered the primary maternal signal for recognition of pregnancy in ruminants such as buffalo, cattle, and sheep (Bazer et al., 1997). In our study, it was found that the IFN-τ expression level was higher in female than male NT blastocysts, as also observed in IVF bovine embryos (Larson et al., 2001). Also, the relative mRNA abundance of IFN-τ was higher in IVF blastocysts than female and male NT blastocysts. This is particularly important, since a higher rate of pregnancy failure is considered the primary reason behind the very low live offspring rate obtained with NT embryos (Chavatte-Palmer et al., 2012).
It has been proposed that the sex ratio in wild animal populations may be controlled by the sexually dimorphic nature of gene expression, such as that of G6PD, as evidenced by the higher growth rate of male embryos than female embryos (Gutierrez-Adan et al., 2000). Out of 591 transcripts that were reported to display sex-specific differences, one-fourth were found to be X linked and as a result, upregulated in females, and the rest were autosomal in IVF mouse embryos (Kobayashi et al., 2006);however, in cattle, one-third of the transcripts expressed were found to be responsible for sexual differences (Bermejo-Alvarez et al., 2010a). This contributes to the skewing of sex ratio in case of IVF embryos in favor of male embryos (Gutierrez-Adan et al., 1996; Wrenzycki et al., 2002). In contrast, we did not find in vitro developmental competence of male and female NT embryos to be different.
However, there were significant differences in the expression level of many important genes listed earlier, including that of IFN-τ, which helps in recognition of pregnancy (Bazer et al., 1997). This necessitates carrying out further studies to compare the conception and offspring rate obtained with male and female NT embryos. This is also important, because OCT4 expression was higher in IVF blastocysts than in male and female NT blastocysts, which had a similar expression level of this gene. In the blastocysts, genes such as OCT4 and NANOG, which are associated with pluripotency, are expressed in the inner cell mass; whereas CDX2 expression is limited to the trophectoderm. It is essential for normal embryonic development that all these genes are expressed in a proper manner. Irrespective of sex, the expression pattern of OCT4 and OCT4-related genes has been shown to be abnormal in clones as compared with IVF bovine embryos (Beyhan et al., 2007).
In conclusion, the present study demonstrates that although male and female NT embryos have similar developmental competence, they differ in their quality, as indicated by the differences in TCN and apoptosis level, and their epigenetic status, as indicated by H3K18ac global level. Also, the expression level of several genes, including X-chromosome-linked (XIST, HPRT, PGK, and G6PD), apoptosis- (CASPASE3), and pregnancy-related genes (IFN-τ), but not that of development-related genes (IGF1R and OCT4), was higher in female than in male cloned blastocysts. The NT embryos differed from their IVF counterparts in terms of lower expression level of OCT4 and IFN-τ but not that of IGF1R.
Footnotes
Acknowledgments
Funds provided by the Dept. of Biotechnology, Ministry of Science & Technology, Govt. of India, and National Agriculture Innovative Project (NAIP) grant to S.K.S. (C 2-1-(5)/2007) and M.S.C. (C-2067 and 075) for the present work are gratefully acknowledged. A.S. and S.K.M. are recipients of CSIR-SRF and ICAR-SRF, respectively.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.