
Abstract
Abstract
Pronucleus-like vesicle formation following premature chromosome condensation (PCC) of the donor cell nucleus is the key event for successful generation of cloned rodents by nuclear transplantation (NT). However in rat cloning, this change is difficult to induce in enucleated recipient oocytes because of their inability to maintain maturation-promoting factor levels. In this study, intact oocytes retrieved from nuclear-visualized H2B-tdTomato knock-in rats were injected with Venus-labeled cell nuclei. Because the incidence of PCC under MG-132 treatment significantly increased with the culture period (0%, 10.8%, 36.8%, and 87.5% at 0, 0.5, 1, and 2 h postinjection, respectively), the metaphase plate of the oocyte was removed 1–2 h after the nuclear injection. The NT-derived rat zygotes (n = 748) were activated with ionomycin/cycloheximide and transferred into temporal host mothers, resulting in the harvest of three blastocysts (0.4%) with Venus fluorescence. Two blastocysts were examined for their potential to commit to NT-derived embryonic stem cells (ntESCs). One ntESC line was established successfully and found to be competent in terms of karyotype, stem cell marker expression, and pluripotency. In conclusion, time-lagged enucleation of visualized oocyte nuclei allows the PCC incidence of donor nuclei and generation of NT blastocysts, and the blastocysts can commit to germline-competent ntESCs.
Introduction
A
Premature chromosome condensation (PCC) and the subsequent formation of multiple pronucleus-like vesicles in the injected donor cell nucleus are the key events for successful generation of cloned mice (Ogura et al., 2000b; Wakayama et al., 1998, 1999). However in rat cloning, this change is difficult to induce in enucleated recipient oocytes because of their inability to maintain maturation-promoting factor (MPF) levels (Ito et al., 2005). Once rat oocytes are placed under in vitro conditions, the oocytes activate spontaneously with a time-dependent decrease of MPF activity (Ito et al., 2005; Ross et al., 2006; Zernicka-Goetz, 1991). Enucleation per se is also one of the factors responsible for decreasing MPF activity in rat oocytes (Ito et al., 2005). Although generation of the first cloned rats was achieved by a sequential donor cell nuclear injection into and metaphase plate enucleation from chemically MPF-controlled oocytes (Zhou et al., 2003), the reproducibility of this NT protocol has not yet been confirmed.
In the present study, recipient oocytes were prepared from H2B-tdTomato knock-in rats (modified to visualize nuclei) to allow removal of the oocyte metaphase plate from NT zygotes in which PCC of the injected nucleus was induced. Blastocysts resulting from the NT zygotes were examined to determine whether they can commit to NT-derived ESCs (ntESCs).
Materials and Methods
Animals
Specific-pathogen-free Crlj:WI and Slc:SD rats were purchased from Charles River Japan (Kanagawa, Japan) and Japan SLC (Shizuoka, Japan), respectively. H2B-tdTomato knock-in rats were generated to visualize cell nuclei (Goto et al., 2015) and maintained by backcrossing with Crlj:WI rats. These rats were housed under controlled lighting (12 h light, 12 h dark), temperature (25 ± 2°C), and humidity (65 ± 5%), with free access to laboratory diet and filtered water. All experiments were approved by the Animal Care and Use Committee of National Institute for Physiological Sciences (Aichi, Japan).
Chemicals and media
Chemicals were purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA) unless otherwise stated. Culture medium used for maintenance of donor ESCs and establishment of ntESCs and parthenote-derived ESCs (pESCs) was N2B27 medium (Ying et al., 2008) containing 1 μM mitogen-activated protein kinase kinase inhibitor (cat. no. PD0325901, Stemgent, Cambridge, MA, USA), 3 μM glycogen synthase kinase 3 inhibitor (cat. no. CHIR99021, Axon Ligands, Groningen, The Netherlands), 1000 U/mL ESGRO® (Millipore, Billerica, MA, USA), and 10 μM forskolin, referred hereafter as 2iF medium (Hirabayashi et al., 2014a). For handling oocytes during NT, mR1ECM (Miyoshi et al., 1995) supplemented with 22 mM HEPES and 5 mM NaHCO3, hereafter referred as HEPES-mR1ECM, was used as the basal medium. HER medium (Ogawa et al., 1971) supplemented with 18% fetal bovine serum (FBS; GIBCO, Auckland, New Zealand) was used for uterine flushing to harvest NT blastocysts.
Preparation of donor cells and recipient oocytes
Male ESCs, previously established from homozygous CAG/Venus transgenic rat blastocysts (Hirabayashi et al., 2014a) and Sertoli cells prepared from homozygous CAG/Venus transgenic male rats at 3 days old according to a previous report (Ogura et al., 2000a), were used as donor cells. Briefly, ESCs at passage 11 or 12 were cultured on mitomycin C–treated mouse embryonic fibroblasts (MEFs) in 2iF medium. Sertoli cells, cryopreserved until use, were thawed, washed, and cultured for 1 week in Dulbecco's Modified Eagle Medium (DMEM; GIBCO) containing 3 μg/mL bovine serum albumin (BSA) and 1 μg/mL porcine follicle-stimulating hormone (FSH). After trypsinization in a 0.25% trypsin/0.6 mM EDTA solution, the smallest cell population (mostly in G0/G1 phase of the cell cycle) was selected by fluorescence-activated cell sorting (FACS; model SH800, Sony, Tokyo, Japan).
For recipient oocytes, heterozygous H2B-tdTomato knock-in female rats (3–5 weeks old) were superovulated by intraperitoneal injections of 300 IU/kg equine chorionic gonadotropin (Asuka Pharmacies, Tokyo, Japan) and 300 IU/kg human chorionic gonadotropin (hCG; Asuka Pharmacies) at an interval of 48–50 h. After 14.5–17 h of hCG injection, oocytes were retrieved from oviductal ampullae and surrounding cumulus cells were removed by short-term culture and pipetting in 0.1% hyaluronidase-containing HEPES-mR1ECM supplemented with 5 μM MG-132.
Generation of NT blastocysts
An aliquot (2 μL) of the donor ESCs suspension was mixed with 8 μL of the HEPES-mR1ECM supplemented with 12% (wt/vol) 360-kDa polyvinylpyrrolidone (ICN Pharmaceuticals, Costa Mesa, CA, USA). The donor ESC nuclei were isolated from the cytoplasm by pipetting and injected into cumulus-free intact oocytes using a piezo-driven micromanipulator (model PMAS-CT150, PrimeTech, Ibaraki, Japan) in HEPES-mR1ECM supplemented with 5 μM MG-132. The injected oocytes were cultured for 0, 0.5, 1, and 2 h in MG-132-containing mR1ECM at 37.0°C in 5% CO2 in air, and stained with 10 μg/mL Hoechst 33342 for 10 min to monitor the incidence of PCC under 360- to 370-nm ultraviolet light.
To examine the developmental capacity of the reconstructed zygotes, the oocyte metaphase plate was removed within 30 min under the tdTomato fluorescence (510- to 560-nm excitation light) in HEPES-mR1ECM supplemented 0.5 μg/mL cytochalasin B after temporal culture in MG-132 containing mR1ECM for 1–2 h following injection of either ESCs or Sertoli cells. The reconstructed oocytes were activated artificially with 5 μM ionomycin in mR1ECM for 5 min and subsequently treated with 5 μg/mL cycloheximide in mR1ECM supplemented with 5 μg/mL cytochalasin B for 4 h at 37.0°C in 5% CO2 in air. After an additional culture for 15–18 h in mR1ECM, all of the surviving zygotes were transferred into the oviducts of pseudopregnant Crlj:WI rats at 0.5 days postcoitum (dpc). Cloned blastocysts were harvested by flushing the uterus at 4.5 dpc and confirmed as NT-derived ones by Venus fluorescence under 460- to 500-nm excitation light. Parthenogenetically activated oocytes served as controls for the 4 days of in vivo culture.
Establishment of ntESCs
NT-derived blastocysts were seeded on MEFs in 2iF medium after removal of the zona pellucida in acidic Tyrode's solution. The growing colonies were picked up and seeded again on newly prepared MEFs. After several passages, the trypsinized ntESCs were resuspended in CELLBANKER® 1 cryopreservation medium (Nippon Zenyaku Kogyo, Fukushima, Japan) and cryopreserved at −80°C within a freezing container (Bicell, Nihon Freezer, Osaka, Japan) until subsequent analyses. Parthenogenetic blastocysts served as controls for establishment of pESCs. Presence or absence of Venus and tdTomato genes in ntESCs and pESCs was confirmed by PCR using primer sets (Table 1) with AmpliTaq Gold® 360 Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to the manufacturer's instructions.
RT-PCR, reverse transcriptase-polymerase chain reaction; bp, base pairs.
Karyotyping of ntESCs as well as donor ESCs was performed by Nihon Gene Research Laboratories (Miyagi, Japan). The expression of some stem cell marker genes (Oct4, Rex1, and rNanog), trophectoderm-specific marker gene (Cdx2), primitive endoderm marker gene (Gata6), and a reference gene (β-actin) was examined by RT-PCR. Briefly, total RNA was extracted from ntESCs, donor ESCs, or rat embryonic fibroblasts (REFs) using an RNeasy® Mini Kit (Qiagen, Germantown, MD, USA). The cDNA was prepared using the SuperScript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) and amplified with TaKaRa LA Taq® (Takara Bio, Shiga, Japan) at 33 cycles at 95°C for 30 sec, 55°C for 30 sec, and 72°C for 60 sec, using primer sets listed in Table 1.
The expression of cell protein markers for stem cells was examined as previously described (Hirabayashi et al., 2014b). Briefly, ntESCs and donor ESCs were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 1 h at room temperature. The cells were incubated in 100% EtOH for 30 min at −20°C, rinsed in PBS and blocking buffer (10% goat serum in PBS), and permeabilized with 0.1% Triton X-100 in the blocking buffer for 1 h at room temperature. Then, the cells were incubated at 4°C with a primary antibody against Oct4 (1:200 for 4 h; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Nanog (1:200 for 4 h; Abcam, Cambridge, UK), Sox2 (1:100 for 3 days; Santa Cruz Biotechnology), or SSEA1 (1:200 for 4 h; Santa Cruz Biotechnology). The samples were stained with Alexa Fluor 546 secondary antibodies for 2 h (1:1000 except for Sox2: 1:500; Invitrogen), counterstained with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS, and enclosed by ProlongGold Antifade Mountant (Thermo Fisher Scientific, Inc.). The digital fluorescent images were taken under a confocal laser scanning microscope (model FV1000-D, Olympus, Tokyo, Japan).
Pluripotency and germ-line competency of ntESCs
Chimeric rats were generated by blastocyst injection of the ntESCs or donor ESCs, as described previously (Hirabayashi et al., 2010). Briefly, 10–15 cells were microinjected into the blastocoelic cavity of host Crlj:WI blastocysts. The reblastulated embryos were transferred into the uteri of pseudopregnant Crlj:WI recipients to allow full-term development to pups. When the pups were found to be chimeric rats by Venus expression, the chimerism of the rats was determined using their peripheral blood cells. Leukocytes isolated by osmotic lysis of erythrocytes were analyzed by fluorescence-activated cell sorting (FACS).
To examine the germ-line competency of ntESCs to the F1 generation, round spermatid injection (ROSI) (Hirabayashi et al., 2008) was applied with a few modifications. Testes of some chimeric rats were microdissected and Venus-positive seminiferous tubules (ST) were selected for ROSI. Round spermatids were sorted by FACS from the testicular cell suspension after a freeze–thaw procedure. Oocytes retrieved from superovulated Slc:SD female rats at 4–5 weeks old were injected with the round spermatids and activated with 5 μM ionomycin in mR1ECM for 5 min followed by an additional 40 min of culture in mR1ECM and 5 μg/mL cycloheximide in mR1ECM for 4 h. The next morning, the ROSI oocytes were transferred into the oviducts of pseudopregnant Crlj:WI recipients, and fetuses at 14.5 dpc were examined for Venus expression.
Statistical analysis
All data except blood chimerism were analyzed by the chi-squared test with Yates' correction using the js-STAR 2.0.7j program (www.kisnet.or.jp/nappa/software/star/index.htm). The percentage data of contribution to blood cells were arcsine transformed and subjected to the Student t-test. A value of p < 0.05 was defined as a criterion of significant difference, except for blastocyst developmental data (p < 0.025 after Bonferroni correction).
Results
The PCC incidence of donor nuclei injected into intact oocytes retrieved from H2B-tdTomato knock-in female rats was examined in the first series of experiments. Both nuclei from donor cells and recipient oocytes were visible in a reconstructed zygote by Hoechst staining (Fig. 1A). The recipient nucleus (metaphase plate of the oocytes) was distinguished from the donor cell nucleus because of the red fluorescence of the H2B-tdTomato gene. The incidence of PCC 30 min after nuclear injection (10.8%, 4/37) was not statistically different from the 0-min value (0%, 0/31), but additional culture up to 2 h resulted in a significant increase of PCC incidence (36.8%, 14/38 at 1 h and 87.5%, 28/32 at 2 h; Fig. 1B). When oocytes were enucleated before nuclear injection of donor cells, PCC of donor nuclei was observed only in 40.0% (12/30) of the reconstructed oocytes even after 2 h of culture. This proportion was significantly lower than the PCC incidence in nonenucleated oocytes at the same culture period (40.0% versus 87.5%).
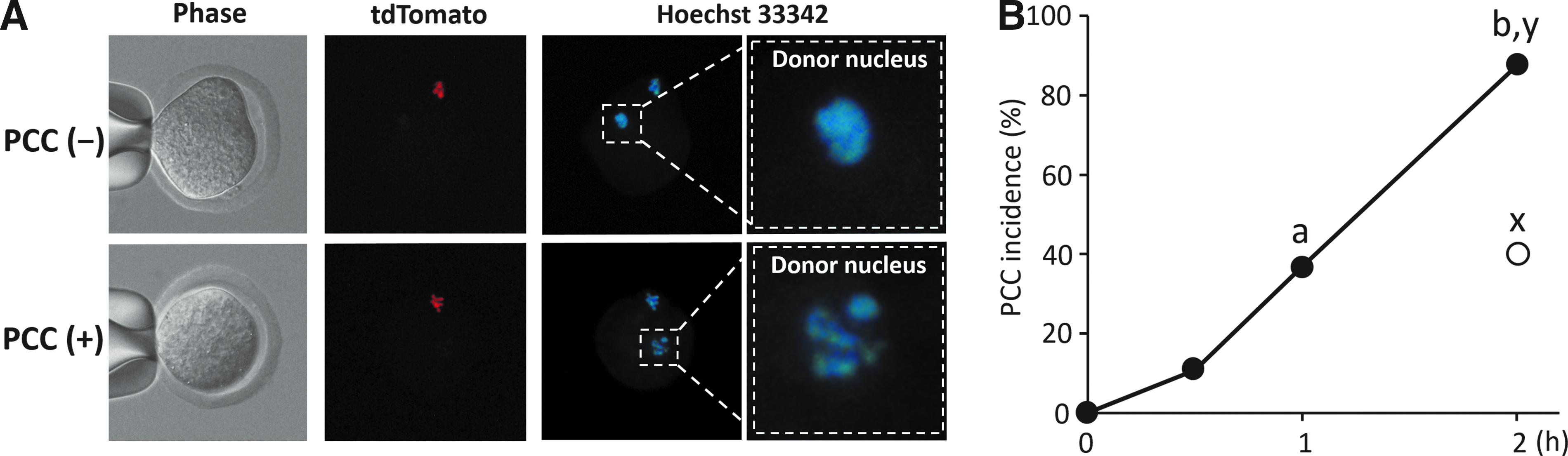
PCC of donor cell nuclei injected into H2B-tdTomato knock-in rat oocytes. (
In the second series of experiments, reconstructed rat zygotes were generated by enucleation 1–2 h following nuclear injection of donor cells (Table 2). Approximately 20% of the oocytes were damaged by nuclear injection and an additional 20% of NT oocytes were damaged by enucleation. Activation treatment with ionomycin and cycloheximide was not detrimental to the reconstructed zygotes. After 4 days of in vivo culture, three blastocysts were harvested from temporal recipient uteri into which a total of 748 reconstructed zygotes derived from ESCs had been transferred. These cloned blastocysts appeared to be morphologically normal, with evidence of Venus-positive and tdTomato-negative fluorescence (Fig. 2). On the other hand, none of 164 reconstructed zygotes derived from Sertoli cells developed to the blastocyst stage. Intact rat oocytes treated with ionomycin and cycloheximide (PA, parthenogenetic activation) could develop into blastocysts at a rate of 37.5% (15/40; Table 2).
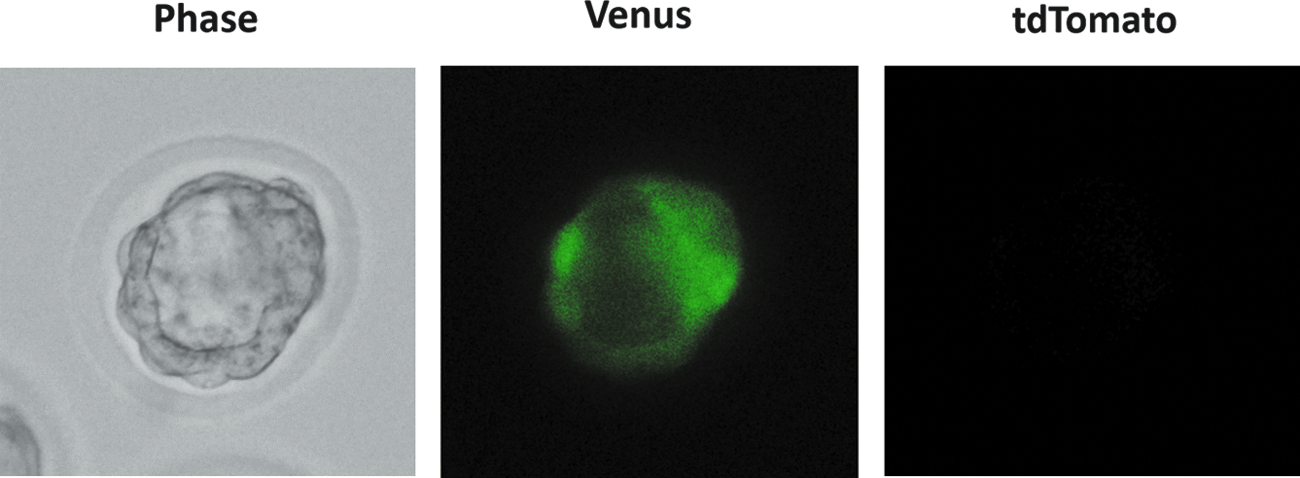
An NT-derived rat blastocyst, harvested from temporal recipient uteri 4 days after transfer of reconstructed zygotes (left panel). The blastocyst is Venus-positive (center panel), but has no fluorescence for tdTomato (right panel). Color images available online at www.liebertpub.com/cell
Different superscripts within a column denote significant difference (p < 0.05).
ntESCs, NT-derived embryonic stem cells; NT, nuclear transplantation; ESCs, embryonic stem cells; PA, parthenogenetic activation.
The harvested blastocysts were subjected to further culture for establishment of ntESCs in the third series of experiments. One ntESC line was established out of two blastocysts seeded on MEFs (Table 2; Fig. 3A) with detection of a PCR product for the Venus gene. Karyotypic analysis confirmed that the ntESCs at passage 7 had a normal karyotype (40XY) in all 50 preparations, as did donor ESCs at passage 12 (Fig. 3B). Expression of stem cell marker genes, such as Oct4, Rex1, and rNanog, was confirmed in both ntESC and donor ESC lines by RT-PCR (Fig. 3C). In addition, a trophectoderm-specific marker gene, Cdx2, was expressed in both cell lines. Expression of marker proteins, such as Oct4, Nanog, Sox2, and SSEA1, was also confirmed in both cell lines by immunostaining (Fig. 3D).
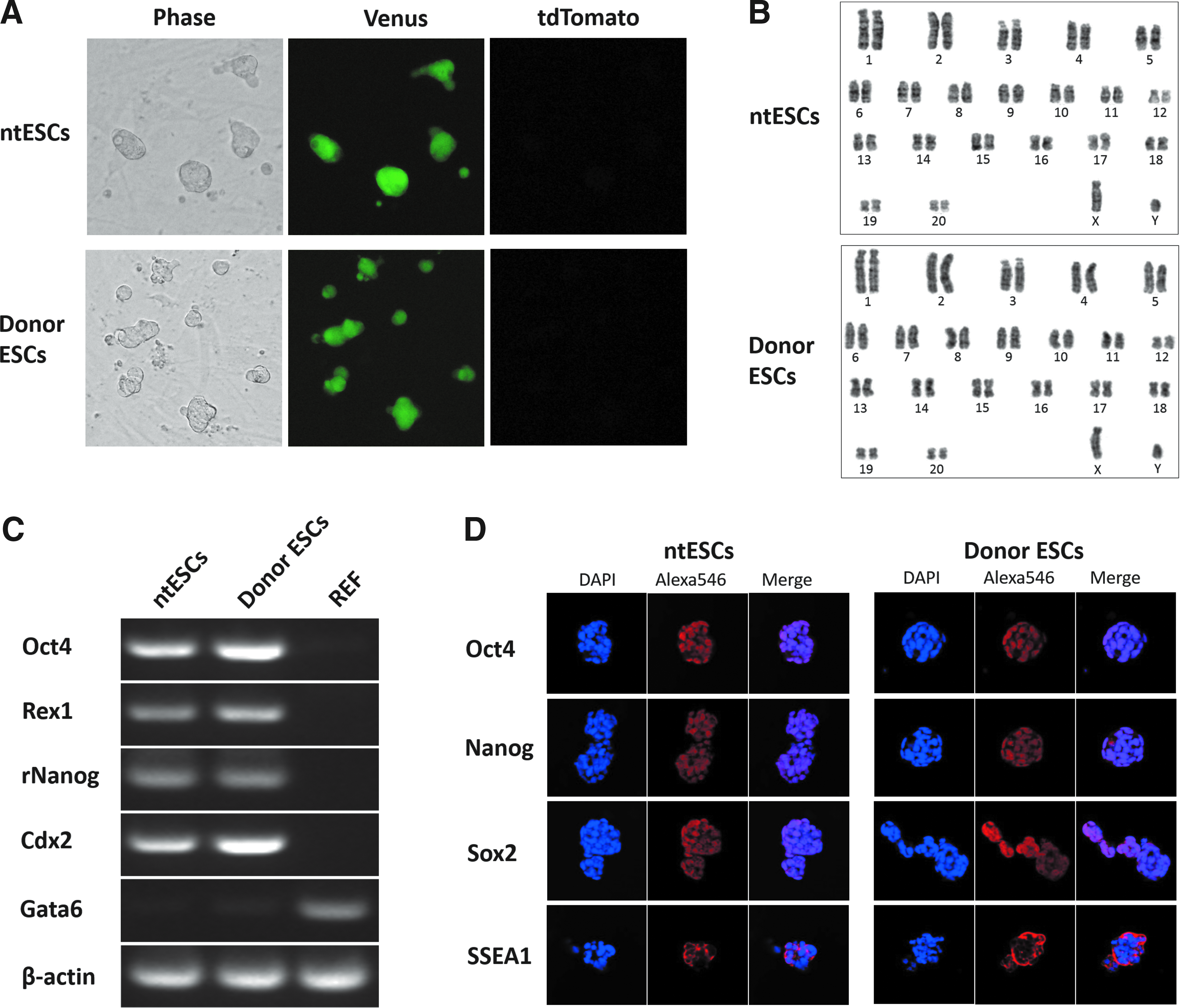
Establishment of rat ntESCs. (
Pluripotency of the ntESC line was examined via chimeric rat production by blastocyst injection in the last series of experiments. Similar proportions of pup birth (35.7% versus 25.0%, respectively) and chimeric rat production (87.9% versus 91.7%, respectively) were obtained after transfer of ntESC versus donor ESC-injected blastocysts (Table 3). The ntESC line contributed to the major organs, including brain, thymus, heart, lung, liver, stomach, pancreas, and kidney, in all chimeric rats, as did the control donor ESC line (Fig. 4A). However, the contribution of the ntESCs into peripheral blood cells was significantly higher than that of donor ESCs (54.4% versus 27.0%, respectively). Proportions of chimeric rats expressing the Venus gene in ST were comparable between ntESCs and donor ESCs (55.6% and 83.3%, respectively; Table 3). Two chimeric rats produced with ntESCs were found to be germ-line competent (Table 3) because Venus-expressing fetal rats were generated by ooplasmic injection of Venus-positive round spermatids (Fig. 4B).
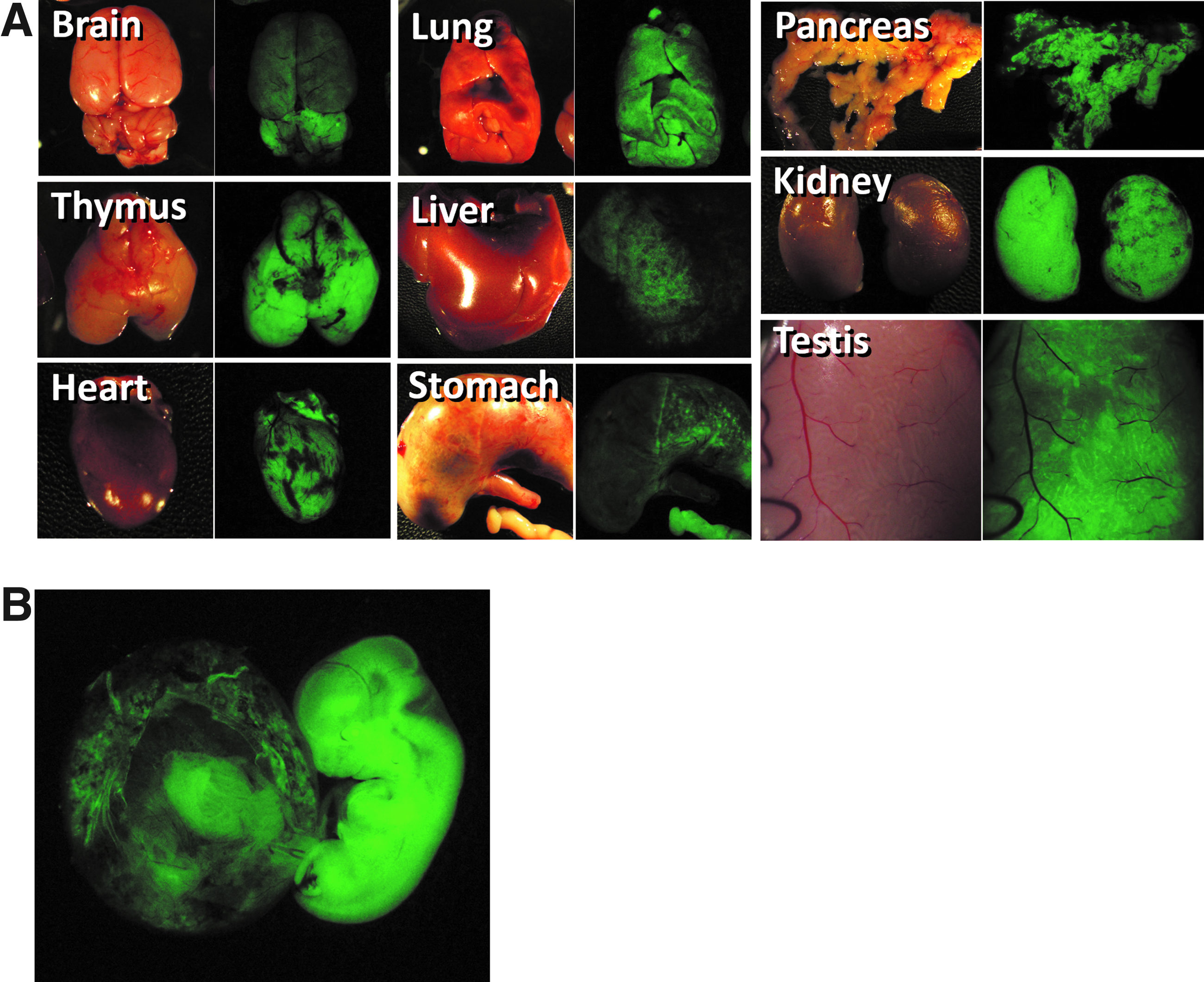
Pluripotency of rat ntESCs confirmed by generation of chimeric rats capable of germ-line transmission. (
Different superscripts within a column denote significant difference (p < 0.05).
ntESCs, NT-derived embryonic stem cells; SD, standard deviation; ST, seminiferous tubule; ROSI, round spermatid injection; GT, germ-line transmission; ND, not determined.
Discussion
A low incidence of PCC in enucleated oocytes may be one of the factors responsible for the low developmental potential of cloned zygotes in rats. This is probably due to decreased MPF activity in spontaneously activating rat oocytes, especially after enucleation of oocyte metaphase plates (Hirabayashi et al., 2003a, b; Ito et al., 2005). Despite treatment with MG132 proteasome inhibitor to suppress spontaneous activation, MPF activity was not maintained in enucleated rat oocytes (Ito et al., 2005). To overcome this obstacle in the present study, rat oocytes were enucleated after injection of the donor cell nucleus. In the first experiment, PCC of donor nuclei occurred at a higher rate in nonenucleated oocytes 2 h after injection (87.5%), which was significantly higher than the PCC rate in enucleated oocytes under MG-132 treatment (40.0%). These results indicate that nuclear visualized oocytes are effective material for rat cloning because they allow a time lag from donor cell nuclear injection to removal of oocyte metaphase plate until PCC of the donor nucleus is induced.
In the present study, we obtained three cloned blastocysts by in vivo culture of rat zygotes reconstructed with ESC nuclei (Table 2). The very low yield of cloned blastocysts in the present study (0.3%) was similar to yields in previous reports (Mizumoto et al., 2008, 2010; Popova et al., 2009), although one publication reported a high blastocyst yield in rat cloning (Hayes et al., 2001). We did not obtain cloned blastocysts using Sertoli cells as nuclear donor (Table 2), and this might be caused by an insufficient number of zygotes reconstructed. A previous study reported that the gene expression profile in mouse blastocysts reconstructed with ESCs appeared to be normal compared with those reconstructed with somatic cells (Kishigami et al., 2006), but the developmental potential to the blastocyst stage is controversial (Riaz et al., 2011; Wakayama et al., 1999). The NT method employed in the present study was effective for inducing PCC, but not enough to support subsequent blastocyst development. The use of Xist knocked-out donor cells (Inoue et al., 2010) or injection of Kdm4d mRNA into NT zygotes (Matoba et al., 2014) may increase the developmental rate, because factors other than the PCC are possibly related to the blastocyst development of NT zygotes.
Because a very limited number of cloned blastocysts could be harvested in the present study, these blastocysts were seeded on MEFs, resulting in the successful establishment of an ntESC line with a normal karyotype and stem cell marker gene expression (Fig. 3). A previous study reported impaired expression of the Oct4 gene in approximately half of mouse ntESC lines (Boiani et al., 2002). Use of ESCs as donor cells for rat cloning in the present study might enhance gene expression, as reported in mouse cloning (Kishigami et al., 2006). Expression of the Cdx2 gene, a trophectoderm-specific marker gene, is a unique characteristic detected in rat stem cells (Hirabayashi et al., 2014b; Hong et al., 2012).
The established ntESC line was able to contribute to the production of chimeric rats at a high rate (87.9%), which was similar to the rate when the donor ESC line was used (91.7%; Table 3). However, the chimerism in peripheral blood cells was higher in ntESCs than that of donor ESCs (54.4% and 27.0%, respectively). Hikichi et al. (2007) reported that the low contribution of mouse pESCs to chimerism was improved by establishing ntESCs with pESCs as NT donors. In addition to the known potential of donor ESCs for germ-line transmission (Hirabayashi et al., 2013), ntESC lines also showed germ-line transmission competency (Fig. 4B). These results suggest that rat blastocysts cloned by nuclear injection and time-lagged enucleation were developmentally normal, at least at the level of inner cell mass cells. Further research is required to examine the potential of development of NT-derived rat zygotes to full-term offspring.
To the best of our knowledge, the ntESC line was established here in rats for the first time, subsequent to mice (Wakayama et al., 2001), monkeys (Byrne et al., 2007), and humans (Tachibana et al., 2013). A recent study showed that DNA methylation and transcriptome profiles were comparable between conventionally prepared ESCs and ntESCs in humans, whereas induced pluripotent stem cells maintained residual DNA methylation patterns of original somatic cells (Ma et al., 2014).
In conclusion, the use of recipient oocytes from gene-modified rats for nuclear visualization could facilitate the PCC of injected donor nuclei. Blastocysts developed from NT-derived zygotes had the potential to commit to germ-line–competent ntESCs. These findings contribute not only to fundamental stem cell biology but also to applied research regarding regenerative medicine.
Footnotes
Acknowledgments
The authors thank Mrs. Keiko Yamauchi and Mrs. Mika Douki (National Institute for Physiological Sciences) for their assistance with animal care and preparation. This work was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Sciences (nos. 22300147 and 25290037).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.