
Abstract
Abstract
A new type of mesenchymal stem cells (MSCs) that expresses stage-specific embryonic antigen 3 (SSEA-3) and the mesenchymal cell marker CD105 are known as multilineage-differentiating stress-enduring (Muse) cells. Studies have shown that stem cells in suspension cultures are more likely to generate embryoid body–like stem cell spheres and maintain an undifferentiated phenotype and pluripotency. We separated Muse cells derived from human dermal fibroblasts by long-term trypsin incubation (LTT) through suspension cultures in methylcellulose. The Muse cells obtained expressed several pluripotency markers, including Nanog, Oct4, Sox2, and SSEA-3, and could differentiate in vitro into cells of the three germ layers, such as hepatocytes (endodermal), neural cells (ectodermal) and adipocytes, and osteocytes (mesodermal cells). These cells showed a low level of DNA methylation and a high nucleo-cytoplasmic ratio. Our study provides an innovative and exciting platform for exploring the potential cell-based therapy of various human diseases using Muse cells as well as their great possibility for regenerative medicine.
Introduction
T
Interestingly, a report showed that a special type of MSC expresses stage-specific embryonic antigen 3 (SSEA-3) and CD105 (a mesenchymal cell marker), namely Muse cells (Kuroda et al., 2010). Muse cells belong to the MSC family and they have the properties of MSCs, but they differ from other mesenchymal cells because of their pluripotency. Muse cells also resemble embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) because they express the pluripotency markers SSEA-3, Nanog, Oct3/4, and Sox2 and are positive for alkaline phosphatase (AP). Moreover, Muse cells possess high potencies for self-renewal and can differentiate into cells of mesodermal, ectodermal, and/or endodermal lineages. Muse cells were first obtained from bone marrow aspirates and human skin fibroblasts subjected to cellular stress in 2010 (Kuroda et al., 2010). More recently, it has been reported that Muse cells are also present in human adipose tissue (Heneidi et al., 2013; Ogura et al., 2014; Simerman et al., 2014). In contrast, non-Muse cells in MSCs do not express pluripotency markers and cannot generate cells representative of all three germ layers. They participate mainly in trophic and immunosuppressive effects in mesenchymal tissues.
Muse cells have attracted a great deal of attention as a new pluripotent stem cell type with great potential for medical research and regenerative medicine. Using fluorescently labeled flow cytometry to sort Muse cells requires expensive equipment and complex operating procedures (Kuroda et al., 2013). In this study, we developed a method to isolate Muse cells from human dermal fibroblasts using long-term trypsin incubation (LTT) through suspension cultures in methylcellulose (MC) medium (Kuroda et al., 2010, 2013), and we then characterized those cells.
Materials and Methods
Preparation of adult human dermal fibroblasts
Skin specimens obtained from adult circumcisions were used to culture cells and were immediately immersed in iodine solution for 5 min and washed with phosphate-buffered saline (PBS) supplemented with 400 U/mL penicillin and 400 μg/mL streptomycin. Subsequently, the subcutaneous adipose tissues were removed manually with the help of eye scissors. The remaining tissues, including the epidermis and dermis, were washed again with PBS and cut into small pieces (approximately 0.5 cm ×2 cm). The dermis was separated from the epidermis after treatment with 0.25% dispase (cat. no. 4942078001, Roche) for 2 h. The dermal sheets obtained were treated with 0.25% trypsin-EDTA (cat. no. 25200-056, Gibco, Grand Island, NY, USA) for 30 min to produce suspensions of individual dermal fibroblasts, which were filtered through a 200-μm filter and then centrifuged at 1000 rpm for 5 min. These dermal fibroblasts were resuspended in α-Minimum Essential Medium Eagle Modification (α-MEM; cat. no. M4526, Sigma-Aldrich, St. Louis, MO, USA) containing 20% (vol/vol) fetal bovine serum (FBS) (cat. no. 085-150, Wisent Bio Products, St-Bruno, Quebec, Canada), 2 mM
MC suspension culture
Culture plates were first coated with poly-HEMA [poly(2-hydroxyethyl methacrylate] (cat. no. P3932, Sigma-Aldrich) to avoid attachment of cells to the bottoms of the plates. The coated plates were then air-dried on a clean bench overnight. No ultraviolet (UV) lamp or blower was used while the the poly-HEMA solution was drying. It has been reported that Muse cells can be enriched by treating human dermal fibroblasts with LTT (Kuroda et al., 2010). Therefore, the cells were suspended in 4–5 mL of 0.25% trypsin solution and incubated at 37°C/5% CO2 in 25-cm2 culture flasks (cat. no. 430639, Corning, Corning, NY, USA) for 16 h. After that, many cells were dead, and continuous vortexing and centrifuging was used to eliminate the dead cells. The cells obtained were suspended in MC Medium (cat. no. 04100, StemCell Technologies, Vancouver, British Columbia, Canada) and transferred to poly-HEMA–coated plates. A volume of culture medium equal to 25% of the initial culture medium was added to the plate every 2 days. After culture for 5–7 days, cell clusters were observed.
Adherent culture
Cells grown in MC suspension cultures generated the first-generation clusters. Cycle culture was performed to evaluate the self-renewal property of the cells obtained, which consisted of suspension culture–adherent culture–suspension culture. After 5–7 days of suspension culture, the first-generation clusters were transferred onto culture plates for adherent culture and expansion. After 7 days of incubation of the first-generation clusters in adherent culture, the expanded cells were treated again by LTT and returned to the MC suspension culture to form second-generation clusters.
AP staining
The first-generation clusters were transferred directly to six-well culture plates and grown for 5–7 days. Cells growing outward from the clusters were observed. The clusters and their proliferating cells were washed at least three times (5 min each wash) with PBS. Staining was performed using the Leukocyte Alkaline Phosphatase Kit (cat. no. 86R, Sigma-Aldrich) according to the manufacturer's instructions.
Analysis of DNA methylation levels
Genomic DNA samples were extracted from cells of the first-generation clusters using a Qiagen QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The absorbance values of nucleic acid at A260 and A280 were measured with a UV spectrophotometer; the purity should be 1.6–2.1. DNA samples that passed the bisulfate conversion quality control subsequently were subjected to PCR analysis using primer sets designed to amplify regions of interest, such as the Oct4 and Nanog promoter regions. Modified DNA by bisulfate conversion was used as a template for PCR amplification, and the PCR products were subcloned and then sequenced by the Shanghai Biological Engineering Co., Ltd.
Bisulfite sequencing was used to detect the methylation levels of the Oct4 and Nanog promoter regions in the cells obtained; the parental dermal fibroblasts served as a control.
Electron microscopy
The first-generation clusters were collected and purified by low-speed centrifugation, fixed in 2.5% glutaraldehyde (buffered to pH 7.2 with sodium cacodylate) for 4 h, and then were postfixed in 1% (wt/vol) osmium tetroxide for 1 h. The specimens were dehydrated sequentially using ethanol at concentrations of 30%, 50%, 70%, 80%, 90%, and 100%, each phase for 10 min. The specimens were then embedded in Epon-Araldite and cut into ultrathin sections (70 nm thick) using a Leica Ultracut UCT ultramicrotome (EM UC6, Leica, Wetzlar, Germany), mounted on copper grids and stained with lead and uranyl solutions. Finally, sections were observed using a transmission electron microscope (JEM-2100, JEOL, Tokyo, Japan).
Immunofluorescence staining for pluripotent markers
Two forms of cells were stained—the M-clusters in suspension culture and the M-clusters in adherent culture, which contained the expanded cells that had grown out from the M-clusters. The cells were fixed with 4% paraformaldehyde for 15 min at room temperature, and then were incubated in 0.1% Triton X-100 in PBS for 10 min. After two successive washes in PBS, cells were incubated with a blocking solution containing 5% bovine serum albumin (BSA; Sigma) at room temperature for 60 min. The cells were then stained with primary antibodies specific for Oct4 (1:200; cat. no. ab19857, Abcam, Cambridge, UK), Sox2 (1:200; cat. no. ab59776, Abcam), Nanog (1:100; cat. no. ab21624, Abcam), and SSEA-3 (1:100; cat. no. MAB4303, Millipore, Billerica, MA, USA). All primary antibodies were diluted in 0.1% BSA in PBS and then incubated overnight at 4°C. After washing three times in PBS, these samples were incubated with secondary antibodies in 0.1% BSA in PBS for 2 h at room temperature in the dark, followed by counterstaining with DAPI (1:1,000; cat. no. D9542, Sigma) for 5 min to show nuclei. Isotype-matched mouse antibodies or normal rabbit immunoglobulin G (IgG) were used controls. Staining was observed using a fluorescence microscope (Olympus, Tokyo, Japan).
In vitro differentiation
The first-generation clusters were collected and digested into single cells by treatment with 0.25% trypsin-EDTA. Various differentiation media were used to induce the differentiation of Muse cells to the cell lineages representative of all three germ layers. The inductions were performed according to the manufacturer's instructions of the Human Mesenchymal Stem Cell Functional Identification Kit (cat. no. SC006, R&D Systems, Minneapolis, MN, USA). Muse cells (at a density of 2.0 × 104 cells/cm2) were cultured on collagen-coated dishes in α-MEM supplemented with 10% FBS and an induction supplement for approximately 4 weeks with the culture medium changed every 3 days. The induction supplement for adipose cells contained hydrocortisone, isobutylmethylxanthine, and indomethacin in 95% ethanol, whereas the induction medium for osteocyte cells contained dexamethasone, ascorbate-phosphate, and β-glycerol phosphate.
For neural induction, Muse cells were cultured at a density of 1.0 × 104 cells/cm2 in poly-HEMA–coated plates for 7–10 days in Neurobasal Medium (cat. no. 21103-049, Invitrogen, Carlsbad, CA, USA) supplemented with B-27 (cat. no. 17504044, Gibco), 30 ng/mL basic fibroblast growth factor (bFGF; PeproTech, Rocky Hill, NJ, USA), and 30 ng/mL epidermal growth factor (EGF) (PeproTech) to induce sphere formation. The cell spheres were then harvested and transferred onto collagen-coated plates and cultured for 3 weeks in α-MEM supplemented with 2% FBS, 25 ng/mL basic fibroblast growth factor (bFGF), and 25 ng/mL brain-derived neurotrophic factor (BDNF; PeproTech) for 6 weeks. The culture medium was changed every 3 days. Staining with antibodies specific for nestin (1:200; cat. no. ab176571, Abcam), Neurofilament-L (NF-L; mouse monoclonal antibody, 1:100; cat. no. 13-0400, Invitrogen), and β-III tubulin (1:100; cat. no. ab18207, Abcam) was performed on the induced cells.
For hepatocyte induction, cells at a density of 1.0 × 104 cells/cm2 were cultured on collagen-coated plates in a medium containing Dulbecco's Modified Eagle Medium (DMEM)/F12, 10% FBS, 2 mM glutamine, 10 ng/mL hepatocyte growth factor (HGF; Gibco), and 10 ng/mL epidermal growth factor (EGF; PeproTech). During the first 2–3 days, 100 ng/mL activin A (R&D Systems) was added to the medium. After removing activin A, the medium was supplemented with 30 ng/mL FGF4 (Gibco). When cells reached 80% confluence after 4–5 days, FGF4 was removed from the medium and the cells were cultured with oncostatin M (10 ng/mL, R&D Systems) and dexamethasone (0.1 μM, Sigma-Aldrich) for 5–7 days. Finally, 1× B27 was added to the medium for the next 7–10 days. Staining with albumin (1:25; cat. no. ab135575, Abcam) was performed on these cells.
Results
Enrichment and MC suspension culture of Muse cells
Primary fibroblasts were obtained from adult human dermis and subcultured to the third generation. Morphologically, they were spindle-shaped (Fig. 1A) and displayed a good ability to proliferate. Cells in earlier subcultures, namely within the fifth passage, were used for the enrichment of Muse cells. After exposing the cells to LTT (16 h of trypsin incubation), the vast majority of cells were dead and only a few cells were surviving; these cells were collected and transferred to MC suspension cultures. Within 5–7 days, individual cells began to proliferate and form clusters that were similar to human ESC-derived embryoid bodies (Fig. 1B).
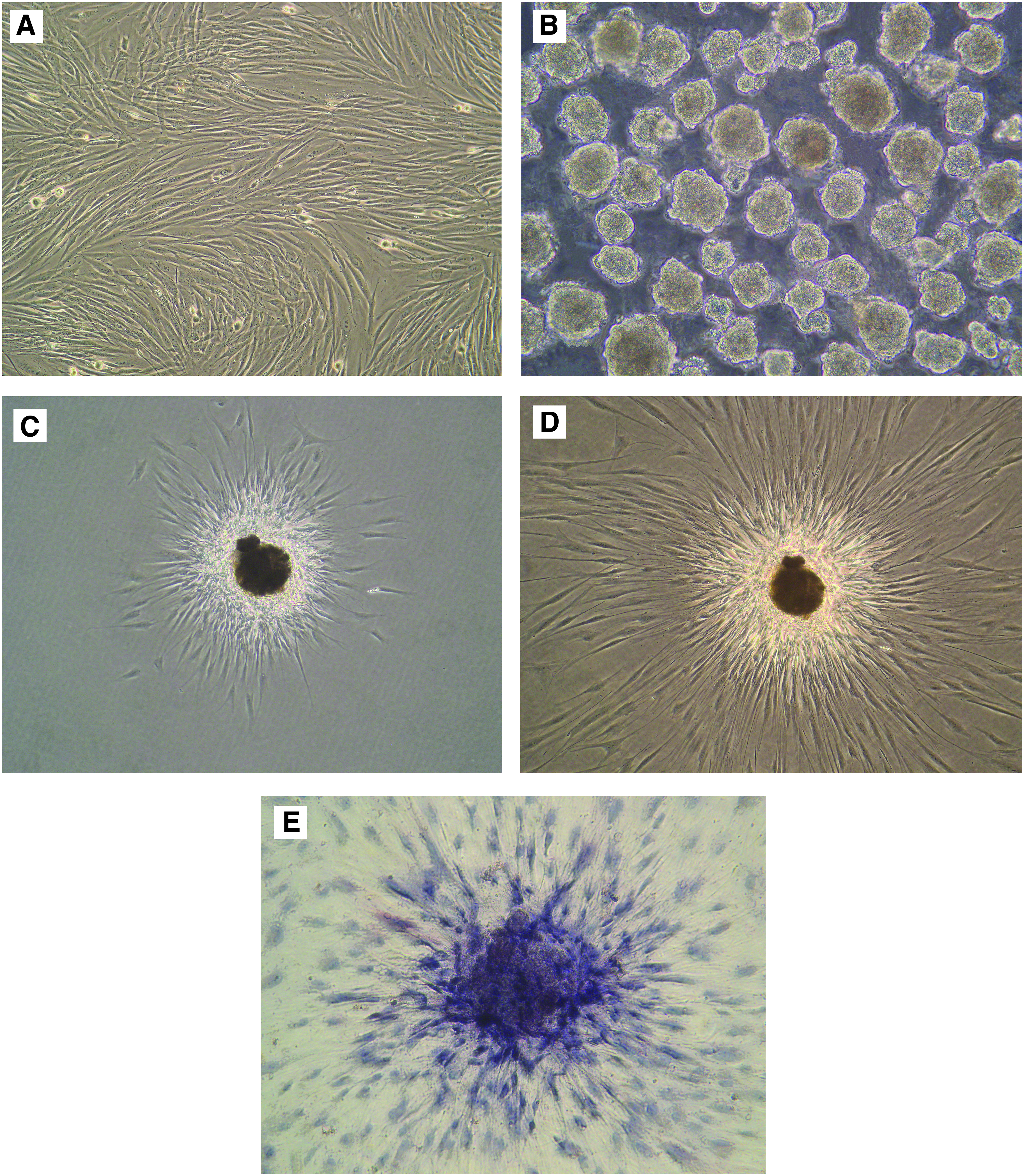
Characterization of Muse cells and M-clusters. (
Expression of AP
Within 5–7 days, most clusters were larger than 25 μm in diameter and were used for adherent culturing and AP staining. When the cell clusters were transferred to culture plates for adherent culture, they quickly attached to the culture plates, after which the cells expanded from the clusters and proliferated (Fig. 1C–D).
AP expression is related to pluripotency (Lu et al., 2011) and is expressed at high levels in undifferentiated cells. AP staining can indirectly reflect the pluripotent state of cells. M-clusters and their expanded cells were positive for AP staining (Fig. 1E). The color was deep in the center of each M-cluster, but gradually faded in cells that expanded around the M-clusters. This indicated high expression of AP in the M-clusters and low expression of AP in cells that expanded from the M-clusters.
Analysis of DNA methylation levels
The DNA methylation sites of M-clusters and dermal fibroblasts in the Oct4 promoter region were 40% and 70%, respectively, and in the Nanog promoter region they were 36.5% and 62.3%, respectively. The difference in DNA methylation level was obvious between the M-clusters and adult fibroblasts. In other words, the results of bisulfite sequencing showed that M-clusters had low DNA methylation levels whereas dermal fibroblasts were highly DNA methylated (Fig. 2).

Comparison of methylation levels between M-clusters and adult dermal fibroblasts. The methylation level in Oct4 promoter regions of M-clusters and adult fibroblasts and in Nanog promoter regions of M-clusters and fibroblasts are shown. The methylation level of M-clusters in Oct4 promoter regions was 40% and in adult fibroblasts was 70%; those in the Nanog promoter regions were 36.5% and 62.3%, respectively.
Electron microscopy
Transmission electron microscopy revealed that Muse cells were more primitive and had a high nuclear–cytoplasmic ratio (Fig. 3A). Some nuclei had a number of processes and a large nucleolus (Fig. 3B), suggesting that those cells may have pluripotent-like features and were undergoing self-renewal. There were few organelles in the cytoplasm, and most were relatively immature, which further illustrated that this kind of cell may not be mature.
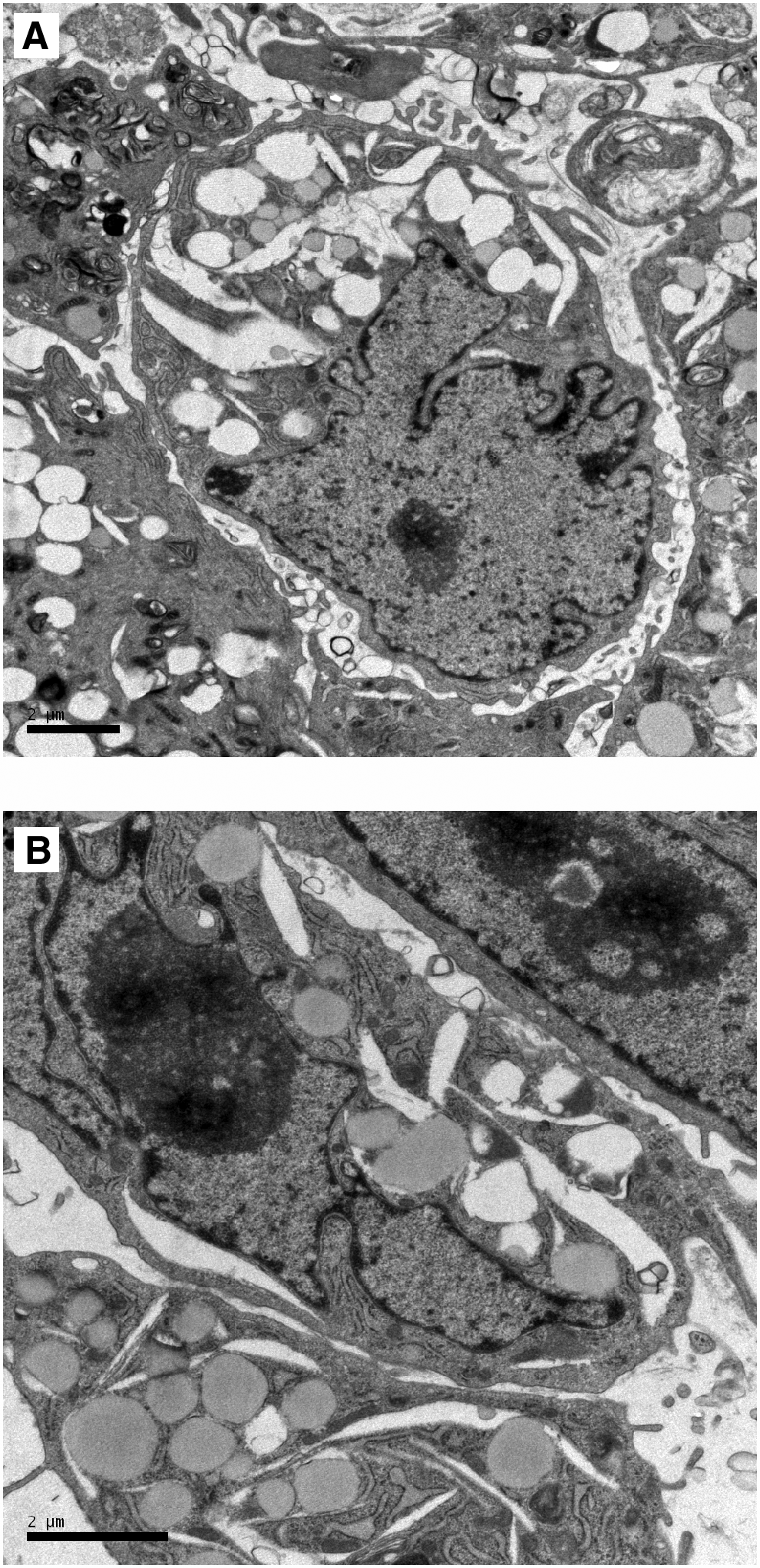
Electron microscopy. (
Immunofluorescence staining of pluripotency markers
It is well known that Oct4, Sox2, Nanog, and SSEA-3 are specific markers that reflect pluripotency (Liu et al., 2013). Immunofluorescence staining showed that the cell clusters were positive for the surface markers Oct4 (Fig. 4A), Sox2 (Fig. 4B), Nanog (Fig. 4C), and SSEA-3 (Fig. 4D) when they were single M-clusters in suspension culture. After 3 days of adherent culture, the cells expanding from the clusters were fixed by paraformaldehyde and were stained by specific antibodies for surface markers, including Oct4 (Fig. 4E), Sox2 (Fig. 4F), Nanog (Fig. 4G), and SSEA-3 (Fig. 4H). The results showed the expression of Oct4 and Sox2 in the M-clusters in adherent culture. However, the fluorescence of Nanog and SSEA-3 was slightly weaker compared to Oct4 and Sox2.
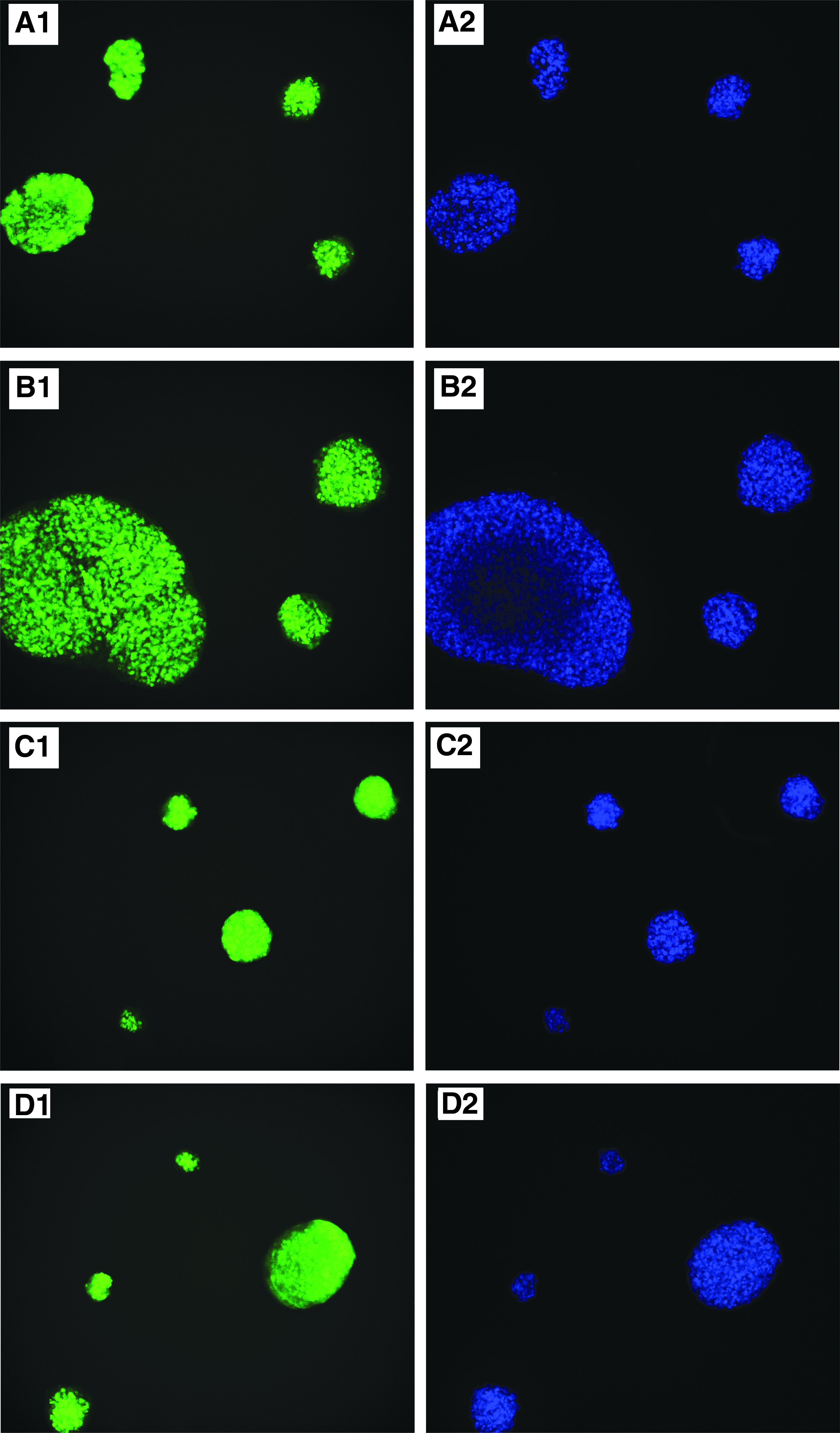
Characterization of Muse cells and M-clusters in adherent culture and/or suspension culture. (
In vitro differentiation
The first-generation clusters were digested into single cells and transferred onto collagen-coated plates. They were then treated with specific cytokines to induce adipocyte and osteocyte (mesodermal), neuronal cell (ectodermal), and hepatocyte (endodermal) differentiation. In adipocyte induction, Muse cells generated cells with lipid droplets that were positive for Oil Red O staining (Fig. 5A, B). Osteocyte induction generated cells positive for Alizarin Red S (Fig. 5C). Neuronal induction demonstrated cells positive for nestin (Fig. 5D), neurofilament (Fig. 5E), and β-III tubulin (Fig. 5F) with a neuron-like morphology. Hepatocyte induction resulted in the generation of cells positive for human albumin (Fig. 5G). Human fibroblasts cultured in the same induction medium were unable to differentiate into cells from any of the three germ layers as a negative control (Fig. 5H).
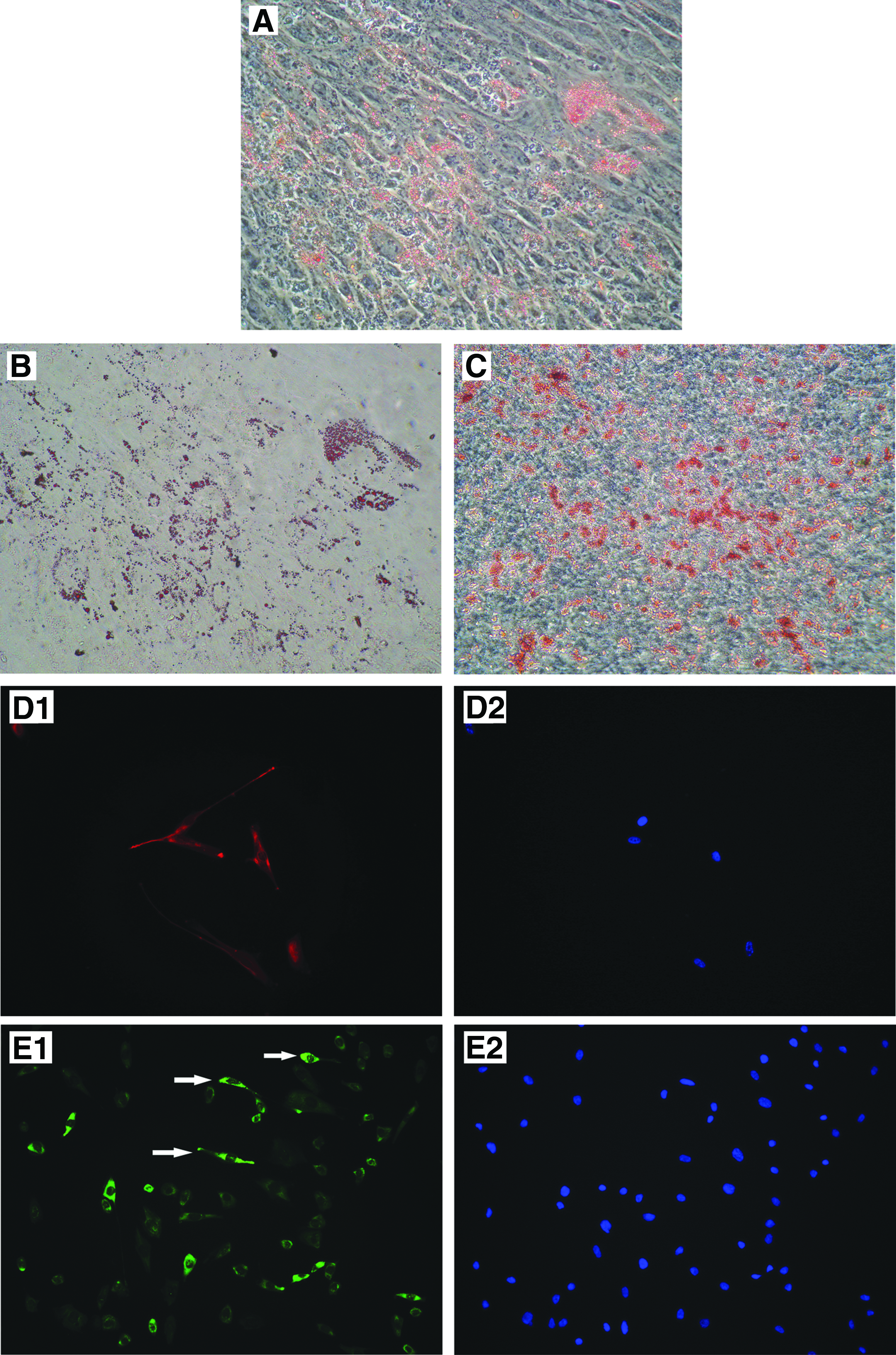
In vitro differentiation of Muse cells into endodermal, ectodermal, and mesodermal lineages. (
Discussion
Muse cells are a peculiar kind of stem cell that is stress tolerant, therefore these cells were named multilineage-differentiating stress-enduring cells. Kuroda et al. performed some stress condition tests, such as poor nutrition, low serum, low O2, repetitive trypsin treatments, and LTT, and reported that a consecutive 16-h trypsin incubation was the most effective for the enrichment of M-clusters in the case of fibroblasts (Kuroda et al., 2010). Some studies indicated that dormant stem cells can be activated under stressful or damaging conditions (Hong et al., 2009; Qiu et al., 2009). Meanwhile, other researchers speculated that suspension cultures may work well for reprogrammed and generated cells that are pluripotent (Chen and Pei, 2012; Fluri et al., 2012). In addition, it was reported that suspension cultures are beneficial to cell expansion and maintain pluripotency (Chen and Couture, 2015; Fluri et al., 2012; Haraguchi et al., 2013).
It had been reported that Muse cells can be isolated from human fibroblasts by fluorescence-activated cell sorting (FACS), but that technique is very expensive and requires complex operating procedures. Therefore, we developed a method to obtain Muse cells through a simpler and more cost-effective approach, namely LTT incubation and culture on MC medium. Using this method, a relatively highly purified population of Muse cells can be isolated without the requirement for FACS. We show that Muse cells have the ability to tolerate stress, in particular LTT, just as their name suggests. Stressful conditions increased the numbers of stem cells. When incubated with trypsin for 16 h, the large majority of dermal fibroblasts die out and only a few cells remain alive. When these surviving cells are cultured in suspension, they form clusters that are similar to human ESC-derived embryoid bodies. The cells obtained show pluripotency characteristics, such as the expression of pluripotency markers, self-renewal ability, expression of AP, and triploblastic differentiation.
The first-generation clusters were positive for pluripotency markers, such as SSEA-3, Nanog, Oct3/4, and Sox2. The second-generation clusters were formed by LTT again after the first-generation clusters were transferred to adherent culture and were expanded. We repeated this cycle of culture, which consisted of suspension culture–adherent culture–suspension culture, to demonstrate the self-renewal and proliferation of the M-clusters. The second-generation clusters showed a similar behavior to the first-generation clusters and remained positive for pluripotency markers and for the expression of AP. Other studies have confirmed that this cycle culture could be performed to the fifth generation and still maintain its pluripotency characteristics (Kuroda et al., 2010). After specific induction, these cells showed positive immunofluorescence staining for human albumin (a hepatocyte marker), nestin, neurofilament, and β-III tubulin (neural cell markers), and also were positive for Oil Red O staining (an adipocyte marker) and Alizarin Red S staining (an osteocyte-like cell marker).
DNA methylation is a stable and heritable marker during early embryonic development and plays an important role in the regulation of gene expression, cell proliferation, differentiation, development, and genomic silencing (Han and Yoon, 2012). In differentiated cells, the Oct4 and Nanog promoter regions are highly methylated and are in an inactivated state, whereas in ESCs and iPSCs, unmethylated or demethylation of these promoters has been observed (Kar et al., 2014; Young, 2011). In Oct4 promoter regions, the methylation level of M-clusters was 40%, but in adult fibroblasts it was 70%. The level of M-clusters and adult fibroblasts in the Nanog promoter regions were 36.5% and 62.3%, respectively.
Bisulfite sequencing showed lower levels of DNA methylation in the Nanog and Oct4 promoter regions of Muse cells compared to non-Muse cells, such as adult dermal fibroblasts. However, the promoter regions of Oct4 and Nanog were partly methylated in Muse cells compared with ESCs and iPSCs. This may explain the pluripotency of Muse cells and their nontumorigenic nature, whereas non-Muse cells do not express pluripotency markers. Ultrastructural analysis of M-clusters revealed a high nuclear–cytoplasmic ratio and few organelles in the cytoplasm, suggesting that these cells are immature. Some investigators have reported that only Muse cells can successfully generate iPSCs (Byrne et al., 2009; Wakao et al., 2013). It is possible that Muse cells will be a viable source for the generation of iPSCs and will contribute to elucidating the mechanisms of cellular reprogramming.
The clinical application of Muse cells has unique advantages (Kinoshita et al., 2015; Wakao et al., 2014; Yamauchi et al., 2015). First, they are an abundant source, are easily derived from mesenchymal tissue, and can be easily purified by LTT or flow sorting using SSEA-3 and mesenchymal cell markers. Second, the proliferation rate of Muse cells is higher; they are self-renewing and have the ability to differentiate into cells representative of all three germ layers. Finally, they can cross the boundaries of the mesodermal lineage and can differentiate into ectodermal or endodermal lineages.
ESCs and iPSCs can form teratomas when transplanted into immunodeficient mouse testes within 8–12 weeks. However, Muse cells do not develop into teratomas when transplanted into the body (Kuroda et al., 2010; Wakao et al., 2011). The nontumorigenic nature of Muse cells may be related to the fact that they reside in mesenchymal tissue. Importantly, this peculiar property, which differs from ESCs and/or iPSCs, brings hope for regenerative medicine and cell-based therapy. For example, when transplanted into the testis of severe combined immunodeficiency (SCID) mice, they can incorporate into damaged areas and can differentiate into tissue-specific cells to repair the damaged tissue (Dezawa, 2011; Wakao et al., 2012). Therefore, the discovery of Muse cells provides new prospects for studying the mechanisms of cellular reprogramming, and they have great potential in regenerative medicine.
Footnotes
Acknowledgments
The authors are very grateful to Professor V.J. Hearing for help with the English-language editing. The work was supported by the National Natural Science Foundation of China (grant no. 81171516).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.