
Abstract
Abstract
Diabetic foot ulcers (DFUs) are nonhealing chronic wounds that are a serious complication of diabetes. Since induced pluripotent stem cells (iPSCs) may offer a potent source of autologous cells to heal these wounds, we studied if repair-deficient fibroblasts, derived from DFU patients and age- and site-matched control fibroblasts, could be reprogrammed to iPSCs. To establish this, we used Sendai virus to successfully reprogram six primary fibroblast cell lines derived from ulcerated skin of two DFU patients (DFU8, DFU25), nonulcerated foot skin from two diabetic patients (DFF24, DFF9), and healthy foot skin from two nondiabetic patients (NFF12, NFF14). We confirmed reprogramming to a pluripotent state through three independent criteria: immunofluorescent staining for SSEA-4 and TRA-1-81, formation of embryoid bodies with differentiation potential to all three embryonic germ layers in vitro, and formation of teratomas in vivo. All iPSC lines showed normal karyotypes and typical, nonmethylated CpG sites for OCT4 and NANOG. iPSCs derived from DFUs were similar to those derived from site-matched nonulcerated skin from both diabetic and nondiabetic patients. These results have established for the first time that multiple, DFU-derived fibroblast cell lines can be reprogrammed with efficiencies similar to control fibroblasts, thus demonstrating their utility for future regenerative therapy of DFUs.
Introduction
D
While our understanding of the pathogenesis of DFUs has increased in recent years (Falanga, 1993; Gurtner et al., 2008; Stojadinovic et al., 2005), it has not been fully exploited to develop new treatments to improve their healing. Existing therapies, such as growth factor treatment (Falanga et al., 1992) and bioactive dressings harboring naïve fibroblasts that do not integrate into host tissues (Dinh and Veves 2006), require repeated therapeutic applications that increase the costs of caring for these wounds (Ehrenreich and Ruszczak, 2006; Mustoe et al., 2006). There is a compelling need to develop alternative cell-based therapies to treat the debilitating complications that result from the inability to heal DFUs (Gurtner et al., 2008; Stojadinovic et al., 2005).
Studies have shown the great potential of cellular reprogramming to generate human induced pluripotent stem cells (iPSCs) as a valuable source of autologous cells for regenerative therapies (Lowry et al., 2008). Previously, we have shown that iPSC-derived fibroblasts trigger a repair-promoting phenotype (Hewitt et al., 2011; Shamis et al., 2011), while others have shown that reprogramming leads to an extended replicative potential (Yehezkel et al., 2011) and improved mitochondrial function (Suhr et al., 2010) when compared with fibroblasts from which they were initially reprogrammed. However, it is unknown if iPSCs that are derived specifically from DFUs can be used to differentiate cells with a biological function that may improve wound repair when these cells are used to treat DFUs.
As a step toward this goal, we demonstrate the first successful reprogramming to iPSCs of primary fibroblast cell lines derived from chronic nonhealing DFUs. In addition to deriving iPSC lines from chronic nonhealing DFUs (DFU8, DFU25), iPSCs were also derived from the nonulcerated foot skin of two diabetic patients (DFF9, DFF24) and from the healthy foot skin of two nondiabetic patients (NFF12, NFF14). These findings provide evidence that DFU-derived fibroblasts can be fully reprogrammed to iPSCs.
Generation of iPSCs derived from fibroblasts harboring multiple phenotypic alterations from diabetic patients now offers new opportunities to model complex diseases when these iPSCs are differentiated into a variety of cell types directly relevant to diabetic complications. Future studies with DFU-derived iPSCs will advance our understanding of how to best activate regenerative functions upon iPSC differentiation that can shift nonhealing fibroblasts to a healing-competent phenotype to ameliorate diabetic complications such as DFUs.
Materials and Methods
Biopsies of DFUs and site-matched diabetic and healthy control patients
Primary fibroblasts were isolated from deidentified skin specimens collected under a protocol approved by the Beth Israel Deaconess Medical Center Institutional Review Board. Patient selection and skin biopsies were obtained after informed consent from healthy subjects (NFF) and patients (DFF, DFU) at Beth Israel Deaconess Medical Center. Three groups of fibroblasts were isolated, including DFU-derived fibroblasts, nonulcerated, site-matched, diabetic foot fibroblasts (DFFs) and nonulcerated, site-matched, nondiabetic foot fibroblasts (NFFs).
Briefly, tissues were incubated overnight at 4°C in 1 mg/mL dispase solution in Dulbecco's modified Eagle's medium (DMEM)/F-12 (Life Technologies) to remove the epidermis. The dermal tissue was minced and incubated in 1.5 mg/mL collagenase solution (Invitrogen) and 125 U/mL hyaluronidase (Sigma) in DMEM/F-12 (Invitrogen) for 1 hour. Red blood cell lysis buffer (Sigma) was added for 2 minutes, and cells were collected by centrifugation.
All fibroblasts were expanded and maintained in fibroblast growth media consisting of DMEM (Life Technologies), 10% fetal bovine serum (FBS; HyClone), HEPES (Sigma), and antibiotic–antimycotic (Life Technologies) at 37°C. Fibroblasts were passaged when confluent using 0.05% trypsin/EDTA and experiments were conducted with cells between passages 4 and 7. Cultures were routinely screened for mycoplasma.
Human fibroblast reprogramming
Reprogramming of fibroblasts from NFF, DFU, and DFF cell lines was performed using the CytoTune®-iPS 2.0 Sendai Reprogramming Kit (Life Technologies). Approximately 1 × 105 human fibroblasts were transferred onto adherent, gelatin-coated tissue culture wells of a six-well plate and cultured with fibroblast medium (DMEM with 10% FBS, 1% HEPES; Life Technologies). The next day, the culture media were changed and the cells were infected with Sendai virus (SeV) at the following multiplicities of infection (MOIs): MOI = 5× for hKOS, MOI = 5× for hcMyc, and MOI = 3× for hKlf4. Twenty-four hours after transduction, the supernatant was aspirated and fresh fibroblast medium was added. Cell culture continued for an additional 6 days and the medium was changed every other day.
Seven days after transduction, fibroblasts were harvested and 1.5 × 105 cells were plated into a six-well plate, which was seeded with a gamma-irradiated mouse embryonic fibroblast (irMEF) feeder layer (R&D Systems). One day after seeding transduced fibroblasts, the medium was changed to iPSC medium containing 80% DMEM/F-12 (Life Technologies), 20% KnockOut Serum Replacement (Life Technologies), 1× Glutamax (Life Technologies), 0.1 mM β-mercaptoethanol (Sigma), and 1% nonessential amino acid (NEAA) stock (Life Technologies). Fully reprogrammed iPSC colonies were transferred to freshly prepared 12-well irMEF plates. iPSC cultures were then maintained on six-well irMEF plates. The medium was changed daily with the addition of 8 ng/mL FGF-2 (ProSpec). Plates were incubated at 37°C, 5% CO2 (Takahashi et al., 2007; Thomson et al., 1998).
Live staining of iPSCs reprogrammed from DFU, DFF, and NFF cell lines
The morphology of reprogrammed iPSCs showed small cells with a large nucleus-to-cytoplasm ratio and well-defined edges similar to human embryonic stem (ES) cells with respect to their size and shape. Analysis of morphological characteristics of iPSCs derived from DFUs (iDFU), DFFs (iDFF), and NFFs (iNFF) was similar regardless of their patient background and revealed tight and flat colonies similar to what has been observed from human ES cells and iPSCs generated from other approaches (Lowry et al., 2008; Park et al., 2008; Takahashi et al., 2007; Thomson et al., 1998).
To characterize colonies generated from SeV-infected skin fibroblasts, we picked an average of 20–30 colonies from each cell line. We utilized a live staining approach (Lowry et al., 2008) to select only the colonies that stained positively for both SSEA-4 (Millipore) and TRA-1-81 (BD Biosciences) antibodies confirming that the iPSCs were fully reprogrammed.
On the day that colonies were large enough to be picked, the supernatant was aspirated and each well was washed once with prewarmed DMEM/F-12. The diluted conjugated SSEA-4 (Millipore) and TRA-1-81 (BD Biosciences) antibodies were added (1:1000) and incubated at 37°C, 5% CO2, for ∼90 minutes. Cells were washed twice with DMEM/F12 and 3 mL of fresh, prewarmed iPSC medium was added to each well. Colonies were evaluated using a fluorescent microscope, and fully reprogrammed colonies were distinguished from partially reprogrammed iPSCs by the presence of both SSEA-4 (Millipore) and TRA-1-81 (BD Biosciences) immunofluorescent staining.
A total of six cell lines, two lines from DFF: iDFF24, iDFF9, two lines from DFU: iDFU8, iDFU25, and two lines from NFF: iNFF12 and iNFF14, were generated. We then chose one clone from each of these lines on which to perform our analysis; iDFF24.10, iDFF9.5, iDFU8.2, iDFU25.10, iNFF12.5, iNFF14.6. A representative of all six clones was then fixed with 4% paraformaldehyde (PFA), stained for pluripotency-associated markers, SSEA-4 (Millipore), TRA-1-81 (BD Biosciences), and OCT4 (Abcam), and counterstained with DAPI (Vector) to confirm that they were fully reprogrammed.
Embryoid body formation
Approximately 10 × 106 iPSCs were used to generate embryoid bodies (EBs) in six-well plates. iPSCs were rinsed with 2 mL Dulbecco's phosphate-buffered saline (PBS; Life Technologies) and enzymatically treated with a mixture of 1 mL collagenase, type IV (1 mg/mL in DMEM; Life Technologies), and 0.5 mL dispase (10 mg/mL in DMEM; Life Technologies) at 37°C, 5% CO2, for 5–7 minutes. When the majority of the colonies detached from the plate as aggregates, they were filtered through a cell strainer (40 μm; BD Falcon) onto a six-well ultralow cluster cell culture plate (Corning) using 15 mL iPSC medium without FGF-2 (2.5 mL/well). Plates were then incubated under standard conditions (37°C, 5% CO2) for 7 days to allow the iPSCs to aggregate into spheroid structures and to prevent adherence to the plate.
From days 2 to 7, half of the medium was replaced with medium consisting of 68% DMEM/F-12 (Life Technologies) supplemented with 1% Pen/Strep (Life Technologies), 15% KOSR (Life Technologies), 15% FBS (heat inactivated, FBS; Hyclone), 1× Glutamax (Life Technologies), 0.1 mM β-mercaptoethanol (Sigma), and 1% NEAA stock (Life Technologies) (Gerami-Naini et al., 2004).
Spontaneous differentiation of iPSCs
We examined the in vitro differentiation of iPSCs using an EB differentiation approach. iPSCs were cultured in suspension using iPSC medium without adding any FGF-2 for 7 days to form EBs. EBs were then seeded onto Matrigel-coated (Corning) plates and cultured with endodermal (STEMCELL Technologies), ectodermal (Life Technologies), or mesoderm differentiation media (EB media as mentioned above). Immunofluorescent staining identified cells positive for the endodermal marker: α-fetoprotein (AFP; Millipore), the ectodermal marker, βIII-tubulin (Millipore), and the mesodermal marker, Vimentin (Abcam), to demonstrate that the iPSCs derived from NFF, DFF, and DFU cell lines (iNFF, iDFF, and iDFU cell lines) have the potential to differentiate into all three germ layers.
Teratoma formation
For the teratoma formation assay, we chose one representative iPSC line from iNFF, iDFF, and iDFU. These cells were cultured on irMEFs and treated with 1 mg/mL collagenase IV (Life Technologies) dissolved in 37°C DMEM (Life Technologies), until detachment of the edges of the iPSC colonies was detected. Approximately 5 × 106 iPSCs in 100 μL DMEM were injected into the rear leg muscles of 5-week-old severe combined immunodeficient (SCID) mice (Taconic). A total of 10 mice were injected.
Mice were sacrificed 8–10 weeks after injection and tissues were excised, washed with PBS, fixed in cold 4% PFA, and processed for paraffin embedding. Sectioned slides were stained by hematoxylin and eosin (H&E) and a variety of cell types representing all three germ layers confirming the presence of teratomas were identified. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Karyotyping
Standard G-banding chromosome analysis was performed for all six lines of iDFF, iDFU, and iNFF at the Cytogenetics Laboratory at Tufts Medical School Department of Pathology and Laboratory Medicine.
Bisulfite pyrosequencing
To evaluate the degree of DNA methylation of the human OCT4 and NANOG promoter in iPSCs, gDNA extracts were sent to EpigenDx and analyzed by bisulfite modification and pyrosequencing analysis of their promoter. Quantitative methylation analyses of six CpG islands in the proximal NANOG promoter were performed through pyrosequencing (EpigenDx) using the ADS502/Human NANOG promoter assay, spanning positions −565 to −431 relative to the NANOG ATG start site (Brakensiek et al., 2007; Tost et al., 2003).
Quantitative real-time polymerase chain reaction (RT-PCR) analysis
For analysis of the presence of SeV genome, RNA was isolated from early (p.3) and late passage (after p.15) iPS cell lines using the Qiagen RNeasy Mini Kit. Five hundred nanograms of RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR (qPCR) was performed using iQ SYBR Green Supermix (Bio-Rad). Cycling conditions were as follows: initial denaturation at 95°C for 3 minutes; 40 cycles of denaturation at 95°C for 10 seconds, annealing at 55°C for 10 seconds, extension at 72°C for 30 seconds; and a final step at 95°C for 1 minute. SeV forward primer was 5′-GGATCACTAGGTGATATCGAGC-3′. SeV reverse primer was 5′-ACCAGACAAGAGTTTAAGAGATATGTATC-3′.
For analysis of the presence of mesenchymal markers in fibroblasts, RNA was isolated from primary fibroblasts and fibroblasts differentiated from iPSCs using the Qiagen RNeasy Mini Kit. Five hundred nanograms of RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad).
qPCR was performed using iQ SYBR Green Supermix (Bio-Rad). Cycling conditions for alpha smooth muscle actin (ACTA2) were as follows: initial denaturation at 95°C for 2 minutes; 40 cycles of denaturation at 95°C for 5 seconds, annealing at 60°C for 30 seconds; and a final step at 95°C for 5 seconds. ACTA2 forward primer was 5′-CATCTCCAGAGTCCAGCACA-3′. ACTA2 reverse primer was 5′-ACTGGGACG ACATGGAAAAG-3′. Cycling conditions for Vimentin were as follows: initial denaturation at 95°C for 3 minutes; 40 cycles of denaturation at 95°C for 10 seconds, annealing at 55.6°C for 10 seconds, extension at 72°C for 30 seconds; and a final step at 95°C for 1 minute. Vimentin forward primer was 5′-ATTCCACTTTGCGTTCAAGG-3′. Vimentin reverse primer was 5′-CTTCAGAGAGAGGAAGCCGA-3′.
Gene expression was normalized to GAPDH. Cycling conditions for GAPDH were as follows: initial denaturation at 95°C for 3 minutes; 40 cycles of denaturation at 95°C for 10 seconds, annealing at 59.2°C for 10 seconds, extension at 72°C for 30 seconds; and a final step at 95°C for 1 minute. GAPDH forward primer was 5′-TGCACCACCAACTGCTTAGC-3′. GAPDH reverse primer was 5′-GGCATGGACTGTGGTCATGAG-3′.
iPSC differentiation toward fibroblasts
iPSC (iNFF, iDFF, iDFU) differentiation toward fibroblasts was performed according to our previously published protocol (Hewitt et al., 2009). Approximately 1 × 106 MEFs were cultured in six-well plates until confluent. MEF cells were then fixed in 4% PFA. iDFU25 cells were passaged at a ratio of 1:6 and cultured in normal human keratinocyte medium containing 3:1 DMEM:F12 (Life Technologies), 5% FCII (Hyclone), 0.18 mM adenine (ICN Biomedicals), 8 mM HEPES (Sigma), 0.5 μg/mL hydrocortisone (Sigma), 10−10 M cholera toxin (MP Biological), 10 ng/mL epidermal growth factor (Austral Biological), and 5 μg/mL insulin (Millipore) (NHK medium). At day 4, 0.5 nM human bone morphogenetic protein-4 (BMP-4; R&D Systems) was added for 3 days.
At day 7, cells were passaged 1:3 onto a fresh fixed MEF feeder layer and cultured in DMEM/F-12 (Life Technologies), 5% FCII (Hyclone), and 1% NEAA and grown for 7 days. At day 14, cells were passaged 1:3 onto a six-well plate (Falcon) without any coating and grown in NHK medium. Cells were expanded on day 21 using type I collagen-coated plates (BD Biosciences).
Flow cytometry
Flow cytometry assay was performed as follows: DFU25 and fibroblasts generated from iDFU25 cells (iDFU25-Fib1) were trypsinized, pelleted, and resuspended in 2% FBS in PBS. The cells were then divided into seven FACS tubes (BD Biosciences) at 2.5 × 105 cells per tube and stained with PE-conjugated anti-CD31, -CD34, -CD73, -CD105, -CD140b, and -IgG1k (BD Biosciences) and unstained. Cells were incubated for 30 minutes at 4°C in the dark and washed with 2% FBS in PBS solution. All data were acquired using FACSCalibur (BD Biosciences) and analyzed using CellQuest (BD Biosciences) and Summit V4.3 software (Dako). Expression was measured in comparison with the corresponding isotype control IgG1k (BD Biosciences) or the corresponding unstained cell samples.
Results
DFU-derived fibroblasts can be reprogrammed to iPSCs
iPSCs were derived using the CytoTune®-iPS 2.0 Sendai Reprogramming Kit (Life Technologies) from three fibroblast cell types, including DFU-derived fibroblasts, nonulcerated site-matched DFFs, and nonulcerated site-matched NFFs that were previously characterized (Maione et al., 2015; Park et al., 2014) (Fig. 1A). Small, flat iPSC colonies with sharp edges emerged after 2 weeks and demonstrated cells with a high nucleus-to-cytoplasm ratio. Colonies were similar in appearance regardless of the origin of primary fibroblasts from which they were derived (Fig. 1B).
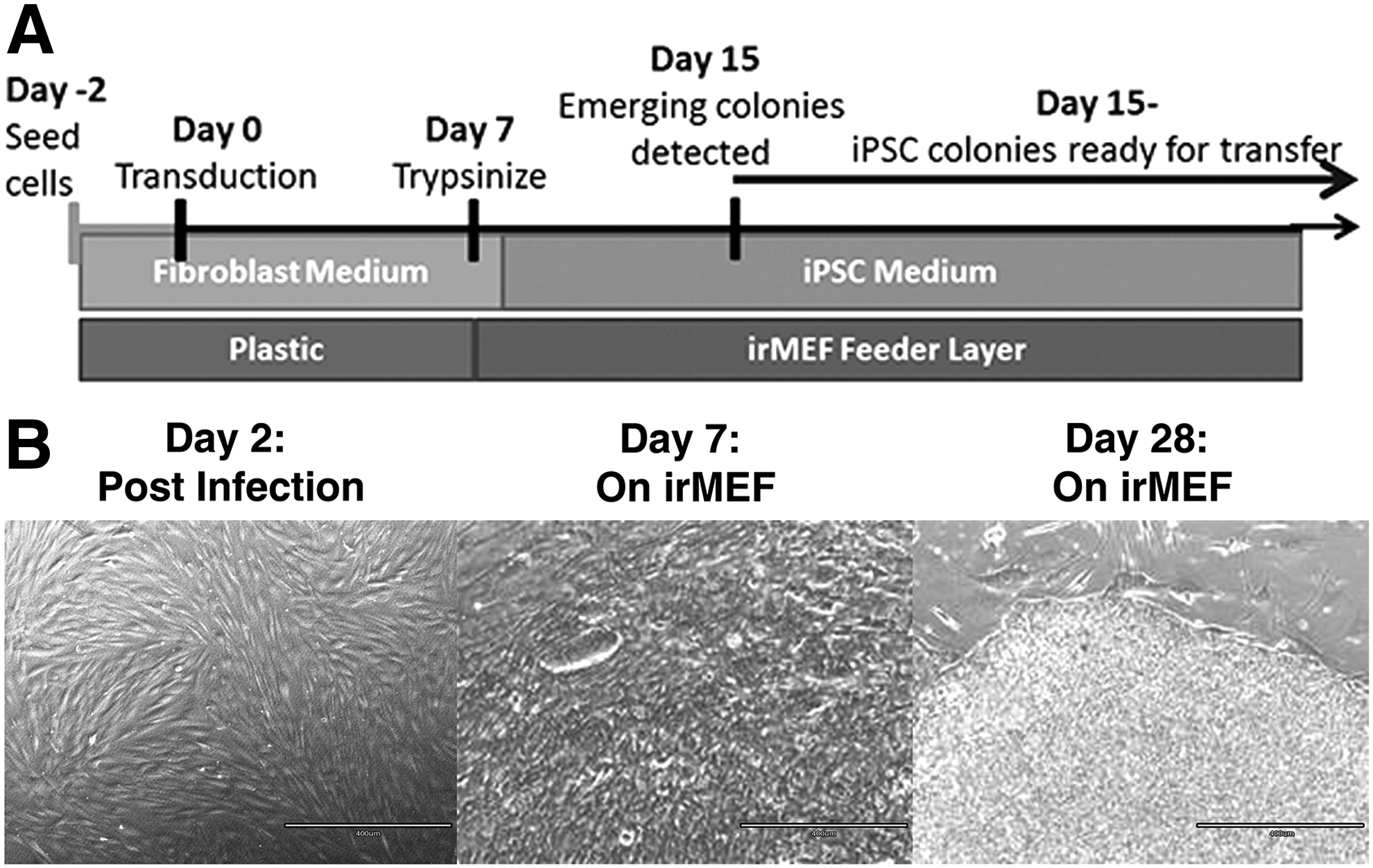
Skin fibroblasts from a representative NFF, DFF, and DFU patient were transduced with the CytoTune®-iPS 2.0 Reprogramming Kit. After 7–10 days post-transduction, cells were transferred onto irMEFs. Colony formation appeared ∼2–3 weeks post-transduction on all six cell lines.
To distinguish fully reprogrammed iPSC colonies from partially reprogrammed iPSC colonies, we performed live cell staining for two pluripotency-associated markers, TRA-1-81 (BD Biosciences) and SSEA-4 (Millipore), as previously described (Chan et al., 2009; Lowry et al., 2008). Figure 2 shows a phase-contrast image of a large fully reprogrammed colony that is positive for both surface markers, TRA-1-81 (BD Biosciences) (green) and SSEA-4 (Millipore) (red). Using live staining as a guide, colonies expressing both Tra-1-81 (BD Biosciences) and SSEA-4 (Millipore) were selected and passaged to generate iPSC subclones that were designated iNFF, iDFF, and iDFU.
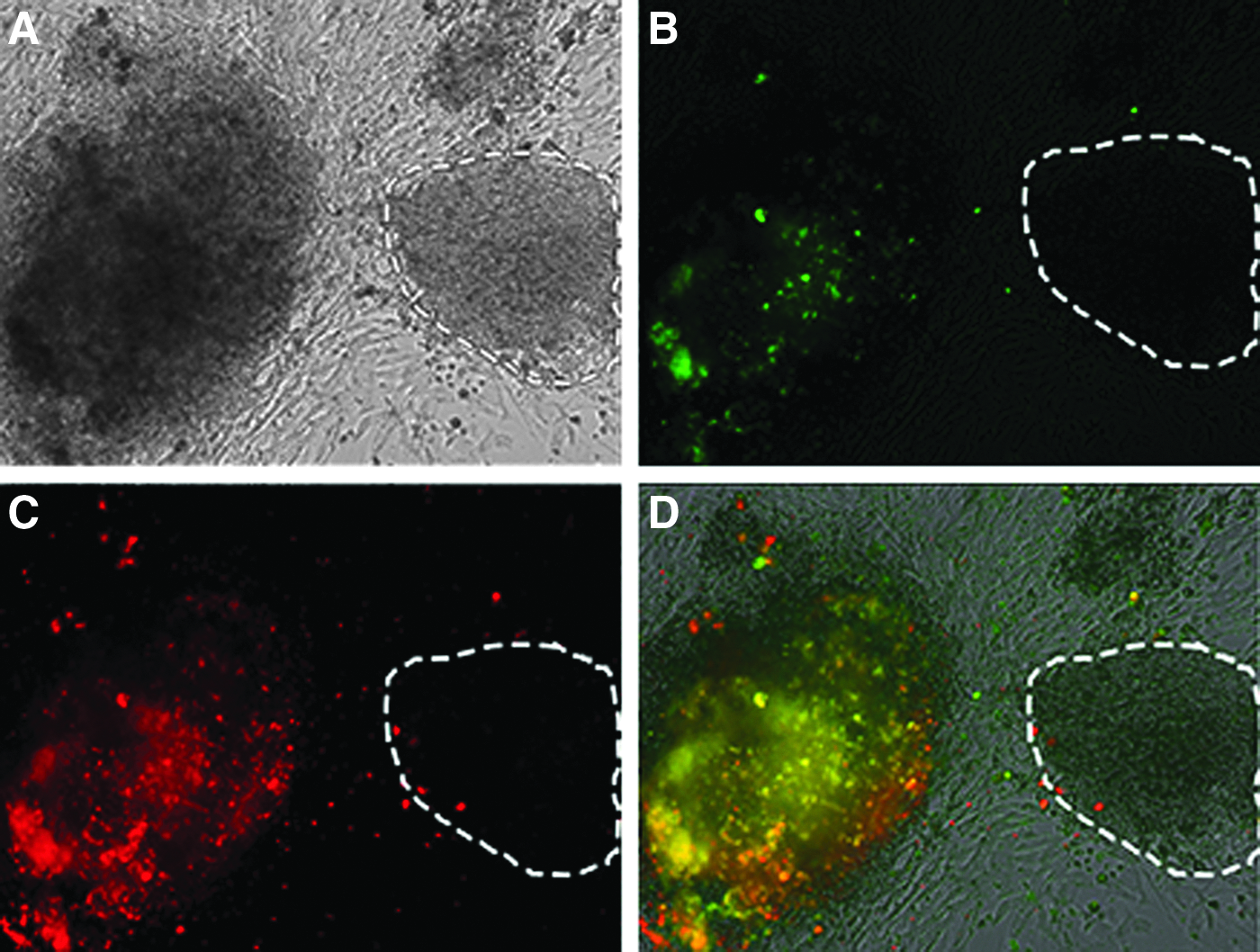
Identifying the fully reprogrammed iPSCs through live staining. The colonies were stained with SSEA-4 (red) and TRA-1-81 (green) to select colonies that were fully reprogrammed.
Overall, efficiencies for reprogramming using SeV in our experiments ranged between 0.33% and 0.64% (Table 1) and were similar for iPSCs reprogrammed from DFUs, DFFs, or NFFs. The iNFF, iDFF, and iDFU subclones were passed every 7–8 days, suggesting that they had acquired similar growth properties.
DFU-derived iPSCs are pluripotent and have a normal karyotype
iNFF, iDFF, and iDFU subclones were then fixed with 4% PFA, stained for pluripotency-associated markers, SSEA-4 (Millipore), TRA-1-81 (BD Biosciences), and OCT4 (Abcam), and counterstained with DAPI (Vector) to confirm that they were fully reprogrammed. Figure 3 shows a representative example of a phase-contrast image of fully reprogrammed colonies that are positive for both surface markers, TRA-1-81 (BD Biosciences) (green) and SSEA-4 (Millipore) (red), as well as DAPI and OCT4 for all six subclones. In contrast, feeder layers of mouse fibroblasts were negative for immunohistochemical staining for pluripotency markers (Takahashi et al., 2007).
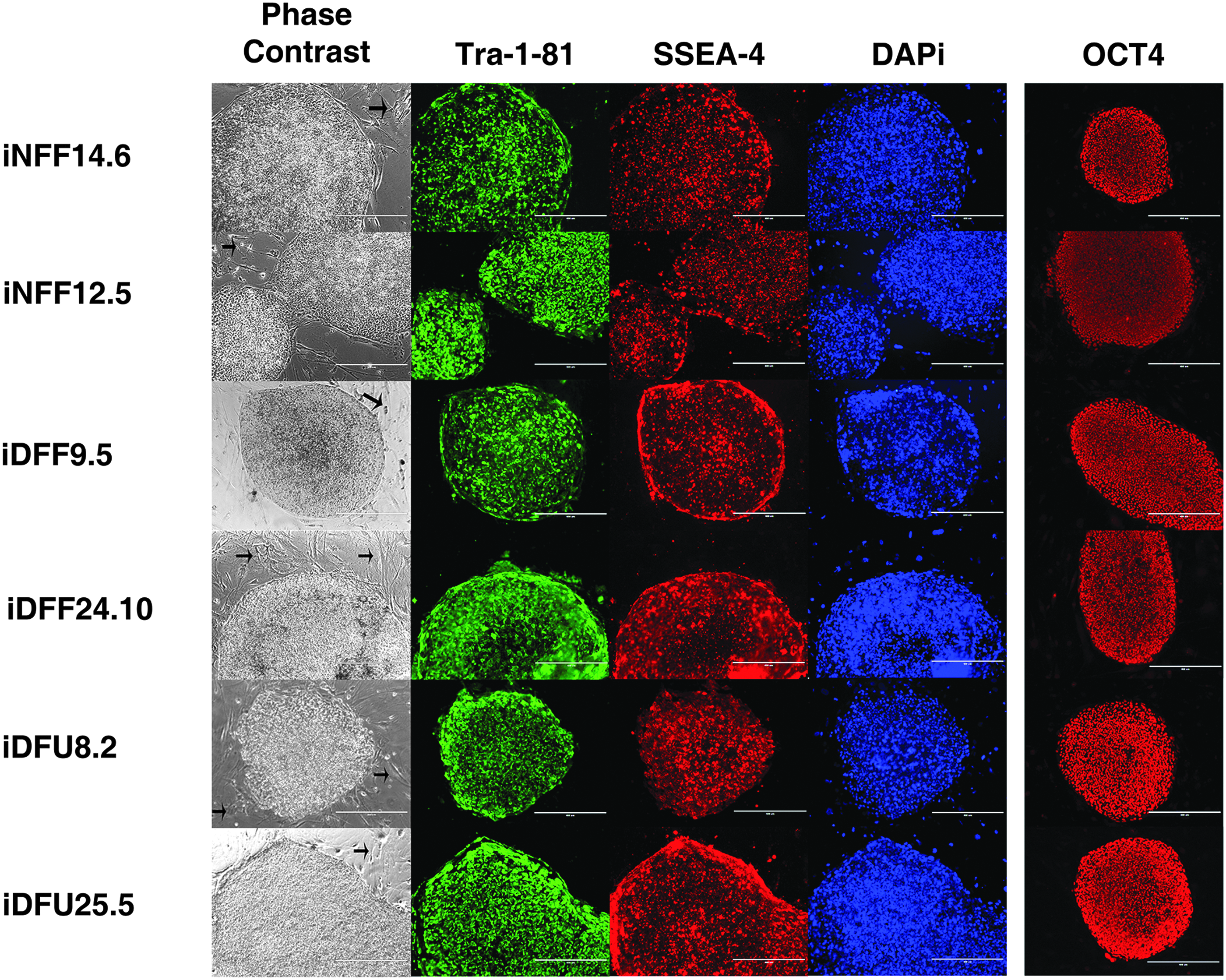
Fully reprogrammed iPSCs from normal human and representative DFF and DFU patients. We show the morphology of iNFF, iDFF, and iDFU cell lines and immunocytochemical analysis of stem cell markers, OCT4, SSEA-4, and TRA-1-81, in these iPSCs. Black arrows in phase-contrast images refer to feeder layers of irMEF, which did not stain with pluripotency markers. Scale bar: 400 μm. iDFF, iPSC derived from DFF; iDFU, iPSC derived from DFU; iNFF, iPSC derived from NFF.
Karyotypic analysis of derived iPSCs revealed that all iNFF, iDFF, and iDFU subclones displayed normal diploid karyotypes (Fig. 4). This karyotypic analysis establishes that two independently derived iDFU subclones that originated from two different patients resulted in a normal karyotype following reprogramming. This was also seen with two independent primary fibroblast lines reprogrammed from the nonulcerated skin of healthy and diabetic patients.
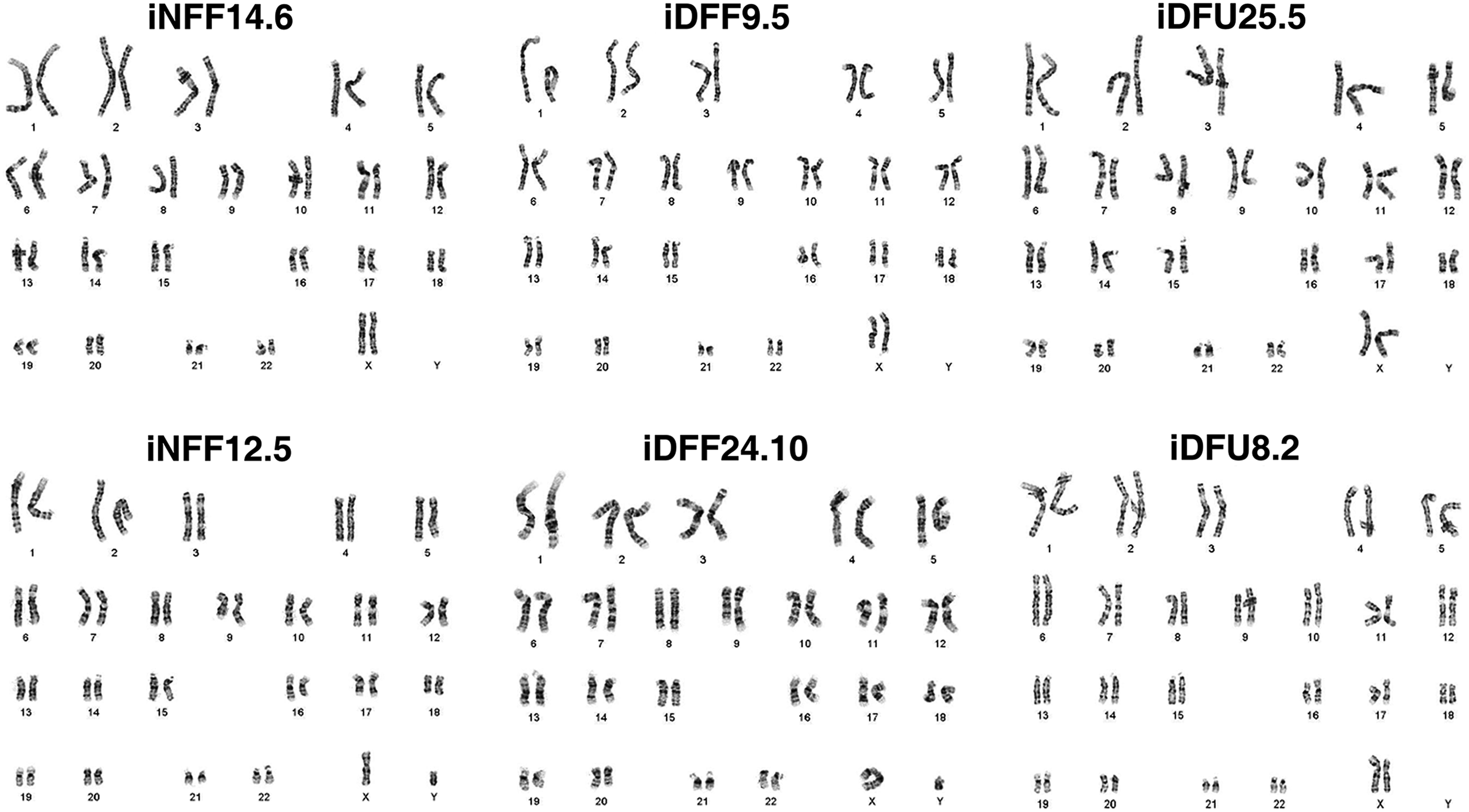
Karyotyping: iNFF, iDFF, and iDFU iPSCs maintained normal karyotype under G-band analysis.
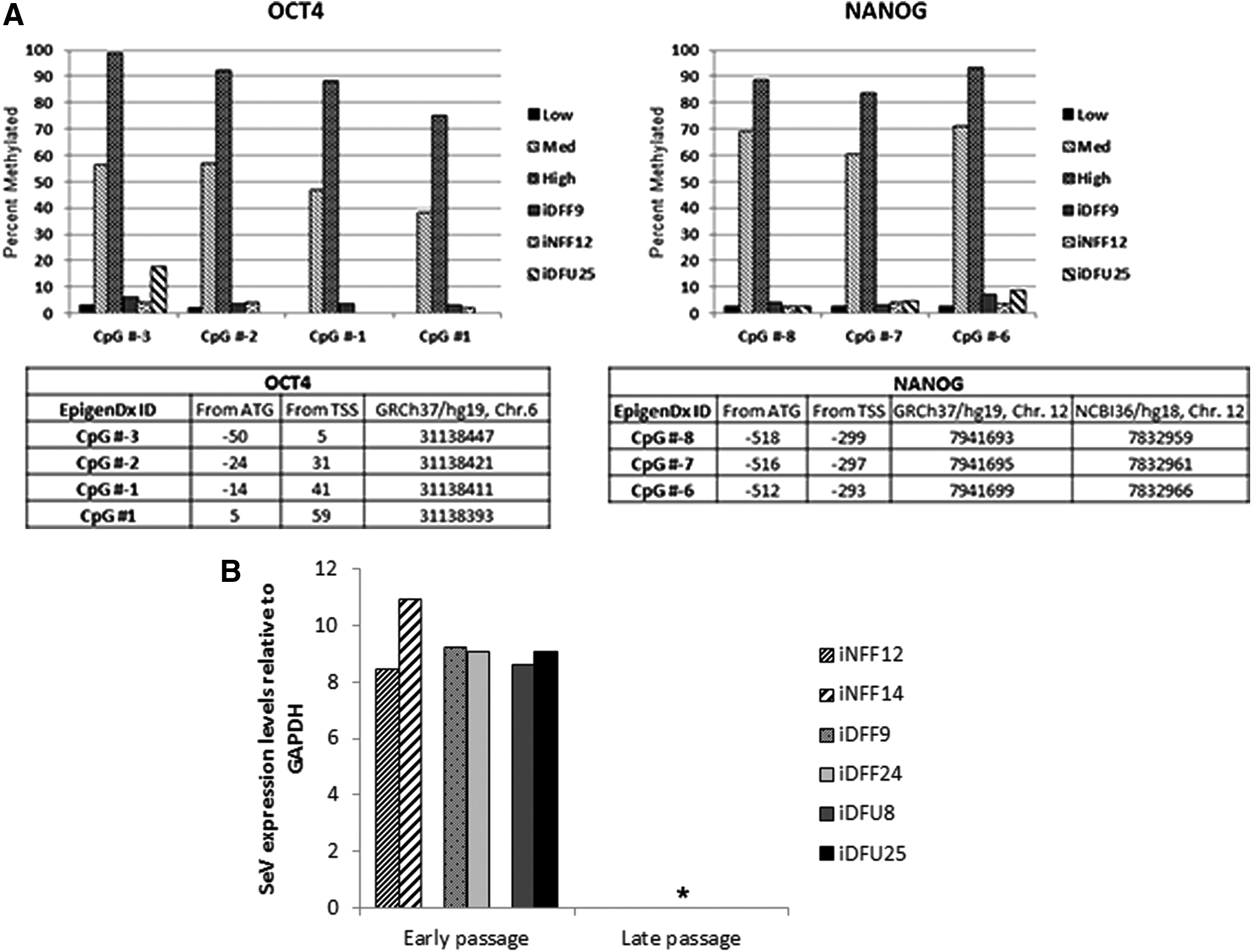
Gene expression and pyrosequencing analyses reveal induction of pluripotency-associated genes through methylation and silencing of transgene expression in iPSC subclones
Since expression and demethylation of promoters of critical pluripotency genes, such as OCT4 and NANOG, occur upon reprogramming, we performed bisulfite pyrosequencing to determine the extent of methylation at the OCT4 and NANOG gene promoters on reprogrammed iPSC subclones for iNFF, iDFF, and iDFU cell lines. DNA methylation analysis revealed that CpG dinucleotides at the OCT4 and NANOG promoter region were demethylated in iDFU25, iDFF9, and iNFF12 subclones (Fig. 5A). This demonstrates that specific CpG sites within NANOG and OCT4 promoters were similarly demethylated regardless of patient history. In spite of expression of the endogenous OCT4 and NANOG genes in iPSCs, quantitative real-time PCR demonstrated that SeV genome was not detected in iNFF, iDFF, and iDFU cell lines at late passages (Fig. 5B). This demonstrates that iPSC subclones are not dependent upon exogenous reprogramming transgenes to maintain their pluripotent state.
DFU-derived iPSCs form EBs and demonstrate normal in vitro differentiation to the three embryonic germ layers
In vitro and in vivo differentiation potentials are the major characteristics of iPSC lines (Takahashi et al., 2007). We assayed the capacity of iPSC subclones to spontaneously differentiate in vitro into all three germ layers by confirming that iPSCs derived from iNFF, iDFF, and iDFU could form EBs. When grown in EB cell culture medium on ultralow adhesive culture plates, iNFF, iDFF, and iDFU cell lines formed EBs within 2 days (Fig. 6A).
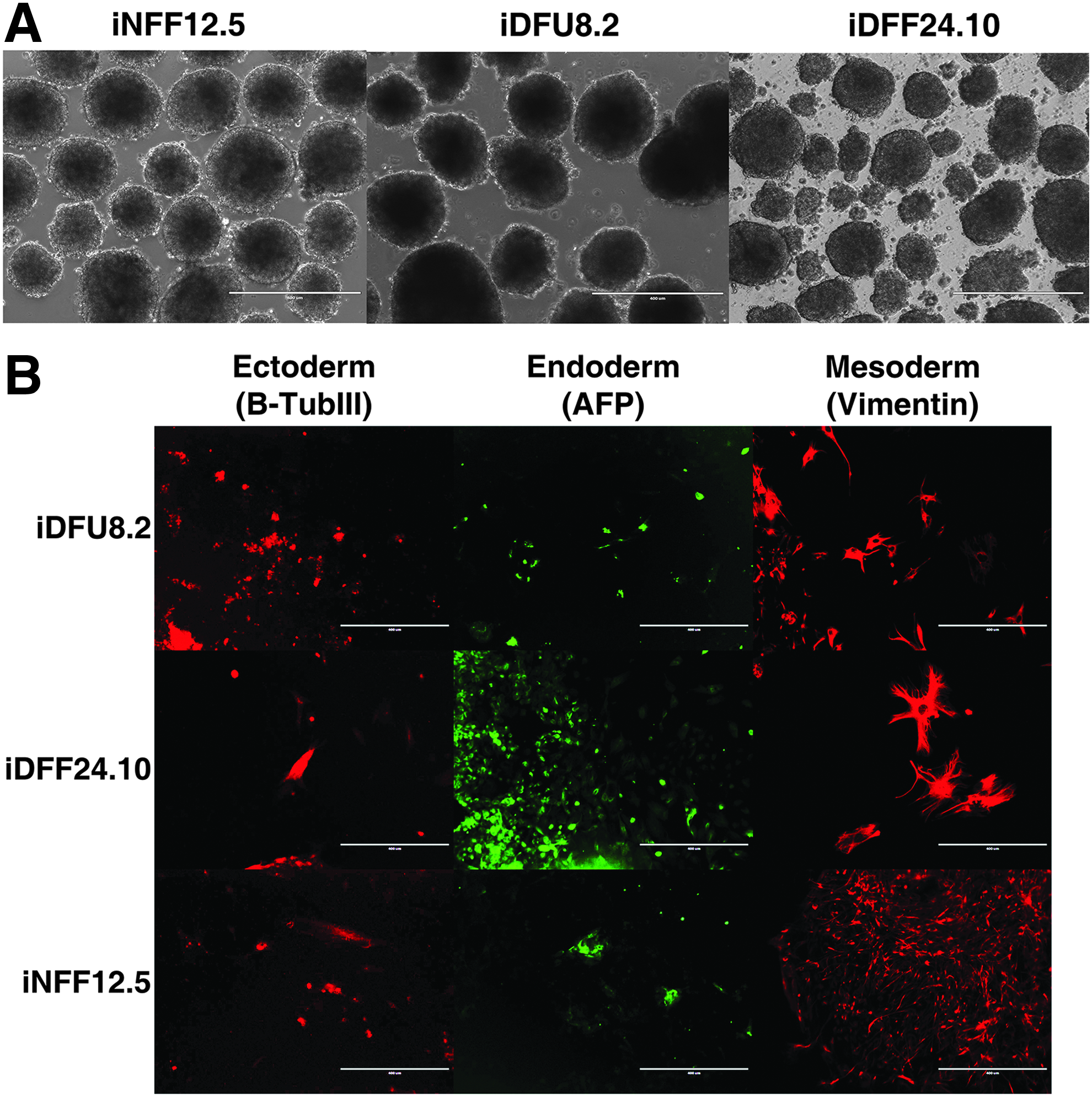
Differentiation analyses of iPSCs: Our results confirmed differentiation capabilities of iPSCs derived from iNFF, iDFF, and iDFU patient into EBs (in vitro approach).
To determine their differentiation potential, we used EB medium (Gerami-Naini et al., 2004) and specialized media to direct cells toward endodermal, ectodermal, or mesodermal fates. Immunofluorescent staining of EBs generated from iNFF, iDFF, and iDFU revealed expression of characteristic markers for βIII-tubulin (ectodermal lineage), AFP (endodermal lineage), and Vimentin (mesodermal lineage) (Fig. 6B). This demonstrated that all iPSC subclones harbor the potential for in vitro differentiation to all germ layers following reprogramming, regardless of their origin.
DFU-derived iPSCs form teratomas in vivo
To test the pluripotency of iPSC clones in vivo, we performed teratoma assays in SCID beige mice. Tumors ranging in size from 1 to 2 cm2 in diameter were excised ∼10–12 weeks after injecting 5 × 106 cells from one representative line of iNFF, iDFF, and iDFU into the dorsal flanks of SCID mice. H&E staining of these tissues revealed teratomas showing tissues representative of all three embryonic germ layers, including cartilage and bands of smooth muscle cells adjacent to glands (mesoderm), intestinal gland-like differentiation with goblet cells (endoderm), and melanin-laden cells denoting neuroectodermal origin (ectoderm) (Fig. 7). These findings confirmed that iPSCs derived from DFUs and site-matched control fibroblasts demonstrated pluripotency in vitro and in vivo.
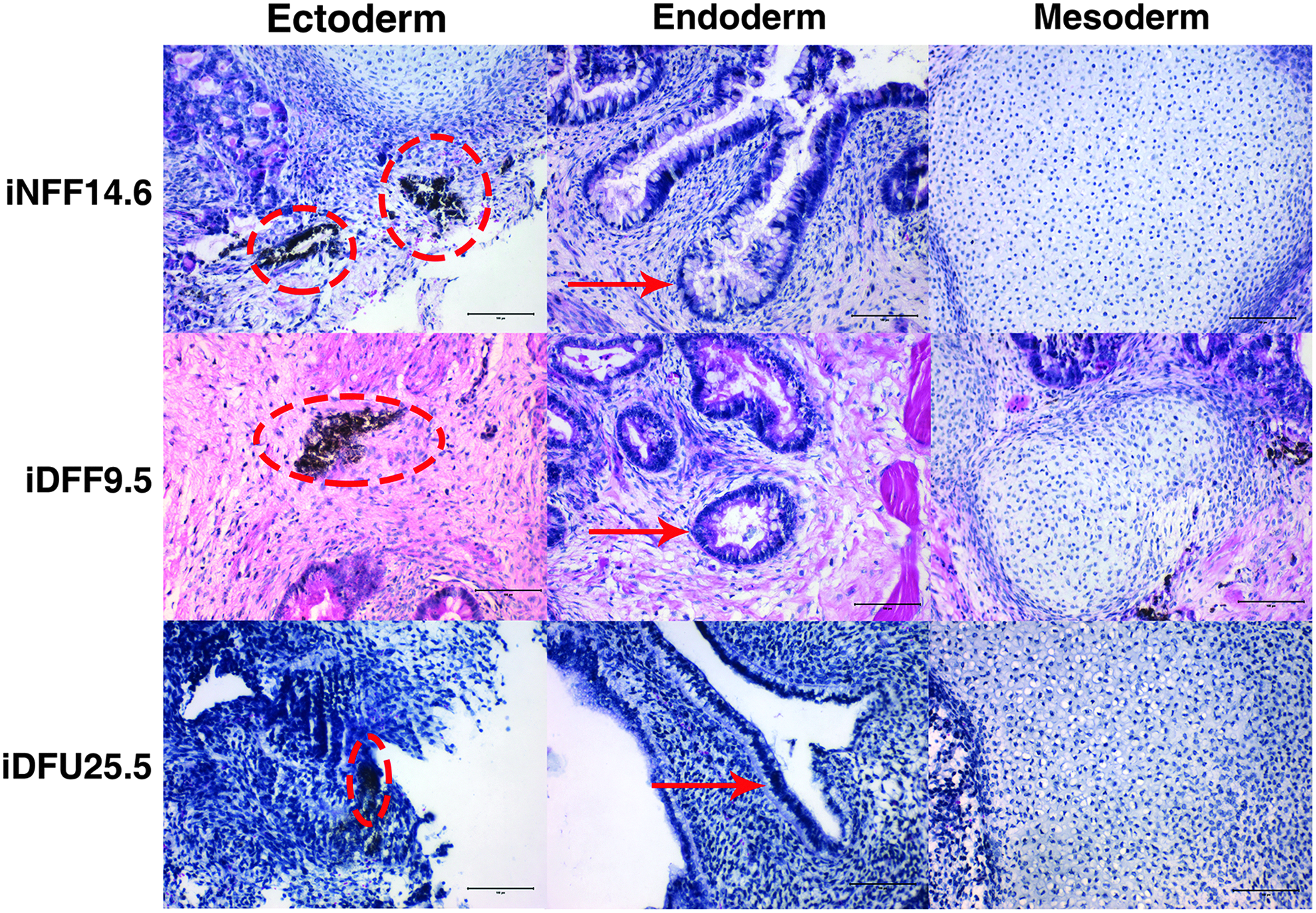
Teratoma formation (in vivo approach): SCID mice were injected with iDFF, iDFU, or iNFF cells intramuscularly. Teratomas were collected, sectioned, and stained with hematoxylin and eosin. Resulting images revealed the following features in ectoderm: melanin-laden cells (marked with red dotted circle) denote neuroectodermal origin; endoderm: intestinal gland-like differentiation with goblet cells; and mesoderm: cartilage. Scale bar: 100 μm. SCID, severe combined immunodeficient.
iPSCs from iDFUs can be differentiated toward fibroblasts
Previously, we have demonstrated that iPSCs derived from human foreskin fibroblasts can be differentiated into fibroblast-like cells (Hewitt et al., 2009). Using this same protocol, we differentiated iPSCs derived from diabetic foot ulcer fibroblasts (iDFU25) into fibroblast-like cells. The resulting cultures, titled iDFU25-Fib1, demonstrate fusiform morphology indicative of mesenchymal fibroblasts (Fig. 8).
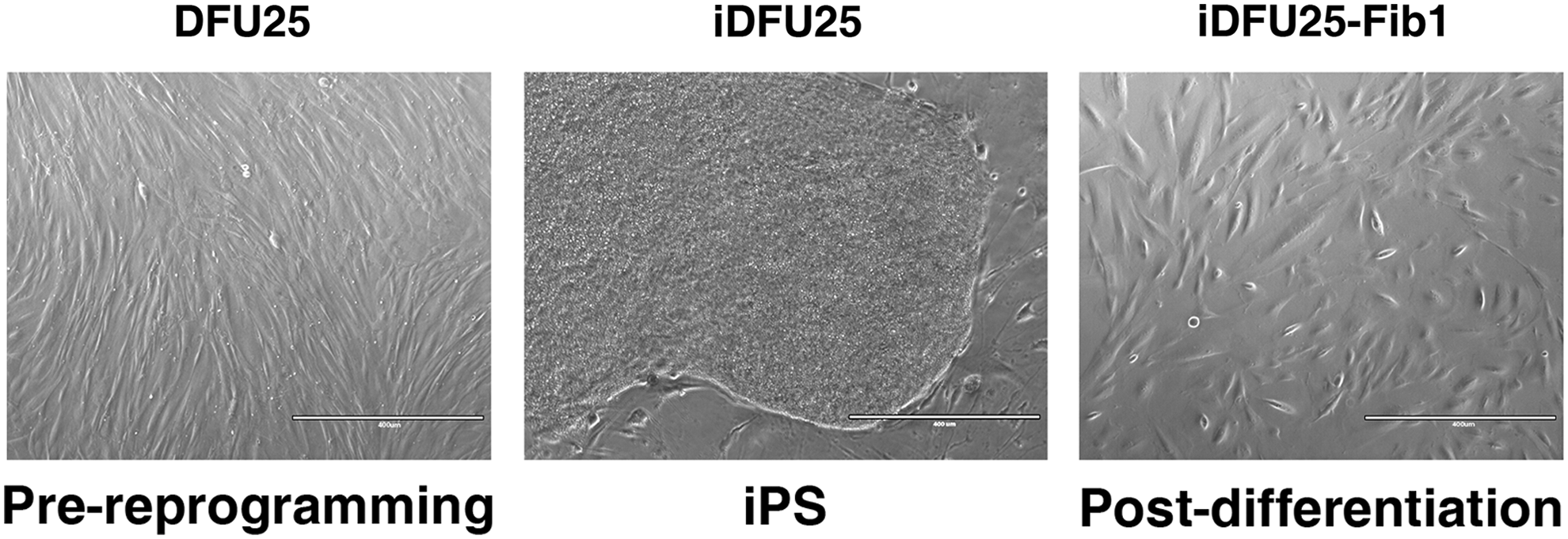
Transition of DFU25 fibroblasts into reprogrammed iPS (iDFU25), followed by differentiation to fibroblast lineage (iDFU25-Fib1). iDFU25-Fib1 exhibits fusiform appearance similar to that of DFU25 and characteristic of mesenchymal fibroblasts.
To further characterize these cells, flow cytometry analysis was performed on DFU25 and iDFU25-Fib1. Expression of mesenchymal fibroblast markers (CD37, CD105, CD140b) was detected as being present in greater than 80% of the cells. Endothelial markers (CD31, CD34) were not detected (Fig. 9A, B) (Hewitt et al., 2009).
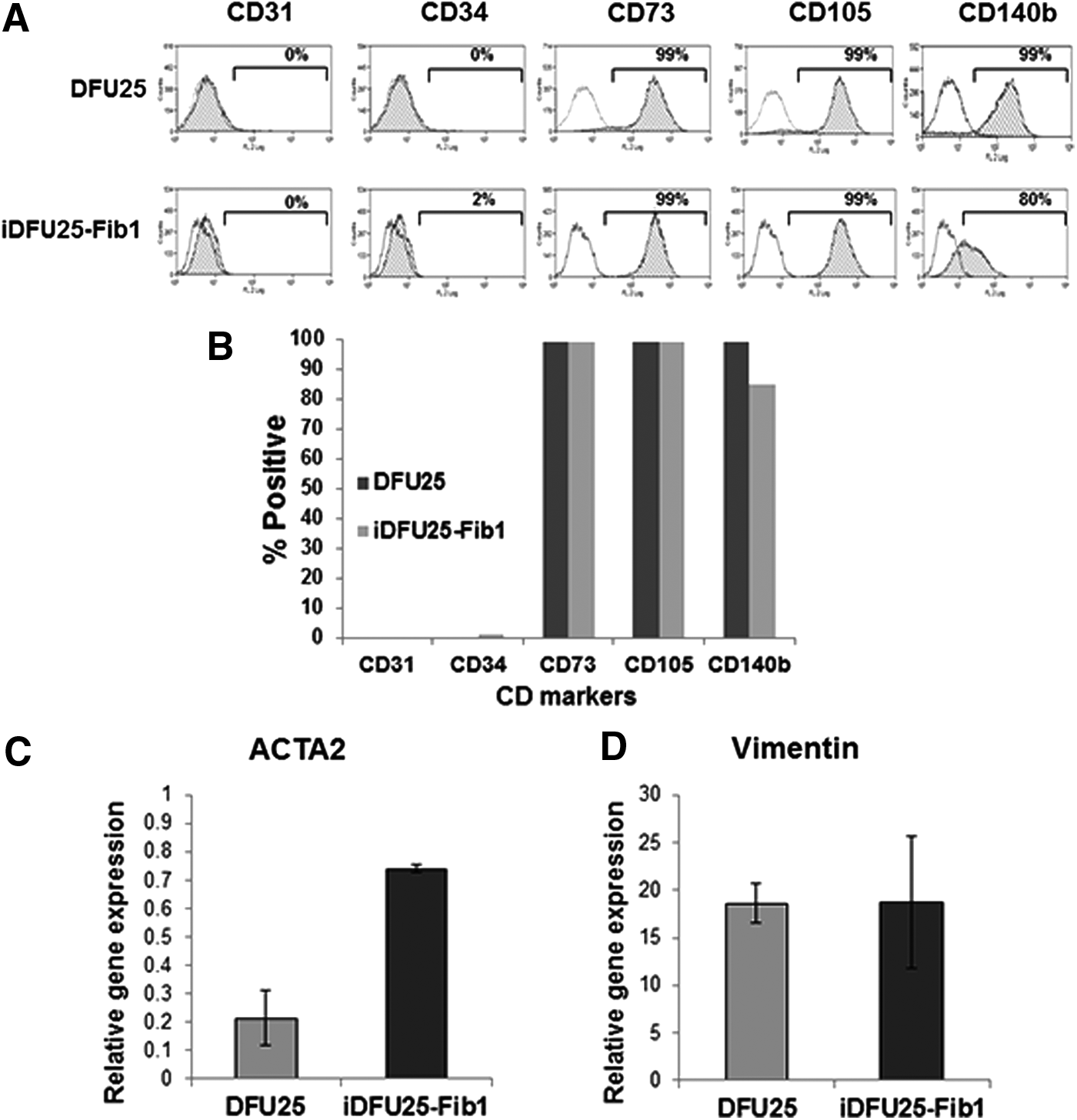
Fibroblasts differentiated from iDFU25 express mesenchymal markers.
Using qPCR, we determined that iDFU25-Fib1 also expressed mesenchymal markers, alpha smooth muscle actin (ACTA2) (Fig. 9C) and Vimentin (Fig. 9D). Taken together, these data indicate that iDFU25, a DFU-derived cell line, has the potential to differentiate into a mesenchymal fibroblast.
Discussion
We have established the capacity to efficiently reprogram fibroblasts from chronic nonhealing wounds from diabetic patients to iPSCs for the first time. By screening and selecting fully reprogrammed iPSC clones from multiple primary cell lines derived from diabetic patients, we provide evidence that reprogramming of DFU cells can be achieved at efficiencies of 0.34%–0.64%. In this study, we utilized SeV to generate these iPSC lines without integrating reprogramming transgenes. The use of this nonintegrating vector system limits the risk for genomic insertions and lowers the likelihood of tumorigenicity for future clinical use in regenerative therapy. Indeed, we determined that exogenous reprogramming transgenes were silenced and were not required to maintain the pluripotent state.
These studies demonstrate the feasibility of reprogramming wound-derived fibroblasts from patients with type 2 diabetes to iPSCs as a first step toward developing strategies to use them to treat these chronic wounds. We also demonstrated the ability of the DFU-derived iPS cells to be differentiated into mesenchymal fibroblasts. Further studies may provide a detailed understanding of whether cellular functions are activated upon differentiation of iPSCs that may reverse wound fibroblasts from a nonhealing to a healing-competent phenotype.
Reprogramming fibroblasts to pluripotency from primary cells derived from DFUs establishes the feasibility of generating cells derived from complex disease sites. There are many examples of reprogramming cell types derived from patients with diseases harboring single gene defects (Zhang et al., 2011). Our studies are the first to reprogram fibroblasts from complex disease environments that manifest multiple cellular defects that prevent cutaneous wound healing. Such cells offer opportunities to learn which cellular functions and regenerative potencies may be restored when cells are differentiated into specific cell types from iPSCs.
Previously our laboratory reported that fibroblasts generated from iPSCs derived from foreskin-derived fibroblasts showed phenotypic and functional features of human stromal fibroblasts (Hewitt et al., 2009) and that these iPSC-derived cells could accelerate epithelial tissue regeneration (Shamis et al., 2011). Future studies will allow us to determine if expression of genes critical to wound repair, angiogenesis, and ECM assembly (Maione et al., 2015; Park et al., 2014), which may be abnormal in DFUs, could be normalized after reprogramming to iPSC and subsequent differentiation to fibroblast lineage.
Previous studies have demonstrated that human iPSCs have been derived from diabetic patients. iPSCs have been reprogrammed from fibroblasts derived from patients with MODY (maturity onset diabetes of the young) for use as in vitro disease models and to explore defects in pancreatic beta cell function and in pancreas and kidney development (Teo et al., 2013). Recently, iPSCs have been generated from fibroblasts derived from patients with insulin receptor mutations to study the genetic basis of insulin resistance in patients with severe insulin resistance due to mutations in the insulin receptor gene (Iovino et al., 2014). We extend these findings by demonstrating that wound-derived fibroblasts from diabetic patients can be reprogrammed with similar properties to those derived from site-matched fibroblasts from healthy skin from diabetic and nondiabetic subjects.
Reprogramming fibroblasts directly from chronic diabetic wound environments provides opportunities to modify the epigenetic profile of these patient-derived fibroblasts for future therapeutic benefits. For example, large-scale epigenetic remodeling, which is known to occur during reprogramming to iPSC (Hewitt et al., 2011), may reverse disease processes under epigenetic control in DFU-derived fibroblasts. Epigenetic remodeling is particularly significant for treating diabetic wounds since metabolic memory, cellular changes induced by hyperglycemia and sustained after return to a normal glycemic state, is regulated by histone modifications (El-Osta et al., 2008; Pirola et al., 2010). These changes alter the expression of genes linked to diabetic complications, including impaired wound responses. Thus, wound fibroblasts reprogrammed to iPSCs offer a powerful model to explore how programs of gene expression linked to the nonhealing phenotype may be shifted to a regenerative phenotype upon the subsequent differentiation of iPSCs to fibroblasts.
Such epigenetic modification could improve the function of diabetic fibroblasts and may reveal controls of genes linked to diabetic complications to offer new ways to study and improve their repair. This could include the capacity to derive a range of cell types from iPSCs originating from diabetic patients to treat diabetic complications at other tissue sites as well.
In summary, we demonstrate that DFU patient-derived fibroblasts can be efficiently reprogrammed to iPSCs and differentiated into fibroblasts. This will lay the groundwork for therapeutic use of these cells directly in the wound bed that may correct cellular repair defects. This will mark a significant departure from the status quo of DFU therapies that will lead the wound repair field to the threshold of novel treatments using autologous cells derived from iPSCs to dramatically improve healing in ways that will significantly reduce long-term care for patients suffering from these conditions.
Footnotes
Acknowledgments
The authors gratefully acknowledge members of their laboratory, Dr. Benjamin Chan for his comments and input on the manuscript, and Ryan Imbriaco and Kamar Reda for their technical assistance. This project was supported by NIH Grant No. R01 DK98055-06 (J.A.G).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.