
Abstract
Abstract
An increasing body of evidence has confirmed existence and function of ovarian stem cells (OSCs). In this study, a novel approach on differentiation of OSCs into oocyte-like cells (OLCs) has been addressed. Recently, different methods have been recruited to isolate and describe aspects of OSCs, but newer and more convenient strategies in isolation are still growing. Herein, a morphology-based method was used to isolate OSCs. Cell suspension of mouse neonatal ovaries was cultured and formed colonies were harvested mechanically and cultivated on mouse embryonic fibroblasts. For differentiation induction, colonies transferred on inactive granulosa cells. Results showed that cells in colonies were positive for alkaline phosphatase activity and reverse transcription-polymerase chain reaction (RT-PCR) confirmed the pluripotency characteristics of cells. Immunofluorescence revealed a positive signal for OCT4, DAZL, MVH, and SSEA1 in colonies as well. Results of RT-PCR and immunofluorescence confirmed that some OLCs were generated within the germ stem cell (GSCs) colonies. The applicability of morphological selection for isolation of GSCs was verified. This method is easier and more economic than other techniques. Our results demonstrate that granulosa cells were effective in inducing the differentiation of OSCs into OLCs through direct cell-to-cell contacts.
Introduction
T
However, some investigators were unable to find these cells and they thereby completely rejected this new discovery (Bristol-Gould et al., 2006; Byskov et al., 2005; Eggan et al., 2006; Gosden et al., 2009; Zhang et al., 2012). The contribution of oogonial stem cells in the reproductive activity of some species such as Drosophila (Waskar et al., 2005; Wong et al., 2005) and teleost medaka (Nakamura et al., 2010) is well established, but the function of these newly discovered cells in adult mammalian ovary has not been completely understood.
Overall, different methods have been employed to establish the ovarian stem cells (OSCs) line (Yazdekhasti et al., 2016). Examples of these methods include magnetic activated cell sorting (MACS), using mouse vasa homolog (MVH) and stage-specific embryonic antigen-1 (SSEA1) antibodies, and fluorescence-activated cell sorting (FACS), using MVH antibody and producing a transgenic mouse model in which green fluorescent protein (GFP) was expressed under a germ cell-specific Oct4 promoter (Gong et al., 2010; Johnson et al., 2005; White et al., 2012; Zou et al., 2009). Moreover, there is one more method that we employed in our laboratory (Khosravi-Farsani et al., 2015; Parvari et al., 2015). In this preplating method, after enzymatic digestion of ovarian tissue, the whole obtained suspension was transferred to gelatin-coated plates. This method has already been used for enrichment of male germ-line stem cells from testis (Guan et al., 2009).
Although there are multiple reports on the isolation and propagation of OSCs, it seems that a more efficient protocol is needed. However, in a study published in Nature Medicine by Tilly group, the isolation and characterization of oogonial stem cells were described comprehensively (White et al., 2012). In 2013, Tilly et al. published a method in Nature protocol and described a detailed isolation procedure for obtaining oogonial stem cells from adult human and mice ovary. Despite this method is fully described and comprehensive, we preferred to apply the simple and efficient preplating method for isolation of OSCs.
Previous studies showed that the presence of gonadal ridge and its somatic cells is crucial for primordial germ cells (PGCs) development through the secretion of different factors that induce differentiation of PGCs into gonocytes (Bukovsky et al., 2005; Geijsen et al., 2004; Lacham‐Kaplan et al., 2006; Mayanagi et al., 2003). Although factors secreted by somatic cells are important for accurate differentiation of PGCs, direct cell–cell contact between somatic cells and gonocytes because of an antiapoptotic effect on gonocytes is also crucial for their differentiation (Brankin et al., 2003). Moreover, it is obviously clear that granulosa cells, as somatic cells compartment of ovarian tissues, have essential impact on oocyte differentiation in vitro and in vivo (Abbasi et al., 2009; Qing et al., 2007). Nevertheless, there is no study or literature that evaluates the impact of granulosa cells on the differentiation of OSCs into oocyte cells in vitro.
In this study, we designed a coculture system between OSCs and granulosa cells from mouse ovary to investigate the effects of granulosa cells on differentiation of OSCs, and morphology-based selection was used as a new strategy to purify OSCs and enrich their colonies as previously described by our group (Parvari et al., 2015).
Materials and Methods
Feeder cell preparation
Previous studies showed that the murine embryonic fibroblast (MEF) feeder layer could facilitate the establishment of mouse OSCs in vitro (White et al., 2012; Woods et al., 2012, 2013); therefore, we used the MEF feeder layer for our initial establishment of OSCs in vitro. For feeder cell preparation, briefly, 13.5-day postcoitus fetuses of CD-1 mouse were sacrificed and the placenta, fetal membranes, visceral organs, heads, and extremities of the fetuses were removed. The embryonic tissue was minced and transferred to an Erlenmeyer flask containing 5 mL of trypsin–EDTA (Invitrogen, Grand Island, NY), a stirrer bar, and a few 5 mm glass beads, then stirred for 15–25 minutes on a magnetic stirrer. The suspension was filtered through a cell strainer and centrifuged at 450 g for 5 minutes at room temperature.
The cell pellet was resuspended and counted using a Neubauer slide. A total of 2 × 106 cells were transferred per 100 mm culture dish in about 3 mL of MEF growth medium (high glucose Dulbecco's modified Eagle's medium [DMEM] supplemented with 15% fetal bovine serum [FBS] and 1% penicillin/streptomycin) and maintained at 5% CO2 and 37°C for 24 hours. After 24 hours, the medium was refreshed to remove debris, erythrocytes, and unattached cellular aggregates and was cultivated for an additional 1–2 days to yield 90% confluence. Afterward, MEFs were inactivated in a medium containing mitomycin C (10 μg/mL) at 37°C for 3 hours. Finally, MEFs were trypsinized and replated on gelatin-coated (0.1%) four-well plates at a density of 50,000–60,000 cells/cm2 (Guan et al., 2006).
Isolation of neonatal OSCs using colony morphology
For isolation of OSCs from ovarian tissue, 20–30 ovaries from 5- to 7-day-old mice were pooled and dissected under a dissecting microscope and collected in ice-cold D-PBS. After centrifugation, D-PBS was discarded and ovaries were transferred to Hank's balanced salt solution without calcium and magnesium containing collagenase (1 mg/mL, Type IV; Invitrogen, Grand Island, NY) and DNase I (10 U/mL, Type I; Sigma, St. Louis, MO) for 45 minutes at 37°C. To speed up dissociation of ovaries, the digestion suspension was pipetted at 10 minutes intervals. The dissociated cells were centrifuged at 400 g for 5 minutes; the cell pellet was resuspended and then passed through a 70 μm nylon cell strainer. A preplate culture for 30 minutes was performed to decrease fibroblast contamination.
Culture of OSCs
After preplating culture, the buoyant cells were transferred onto gelatin-coated 60-mm culture dishes in DMEM supplemented with 0.1 mM β-mercaptoethanol (Sigma, St. Louis, MO), 1% (v/v) nonessential amino acids (Invitrogen, Grand Island, NY), 2 mM
Alkaline phosphatase activity
The alkaline dye was prepared by dissolving fast red naphthol and Tris buffer tablet in 10 mL of deionized water (Sigma, St. Louis, MO). Cells were fixed in 10% formaldehyde for 10 minutes followed by two sequential washing with 0.2 M Tris. Alkaline dye was added to the cells and then incubated for 30 minutes at 37°C; cells were washed and observed using an inverted microscope.
Immunofluorescence
For fluorescent immunocytochemistry, cells were fixed in 4% paraformaldehyde (Merck, Darmstadt, Germany) for 15 minutes and rinsed twice with cold phosphate-buffered saline (PBS) (pH 7.4), followed by permeabilization with 0.2% Triton X-100 (Sigma, St. Louis, MO) for 15 minutes. Nonspecific binding sites were blocked with 1% bovine serum albumin (Sigma, St. Louis, MO) at 37°C for 30 minutes. Primary antibodies against OCT4 (rabbit polyclonal to OCT4; Abcam), SSEA1 (MS MAb to SSEA1 Mc480; Abcam), and germ cell marker DAZL (rabbit polyclonal to DAZL; Abcam) were diluted in blocking buffer and incubated overnight at 4°C. After three washes, FITC-conjugated secondary antibody (goat polyclonal secondary to rabbit IgG, Abcam) (Rb PAb to ms IgG+IgM+IgA FITC; Abcam) was added for 1 hour at room temperature in the dark. Nuclear counter staining was carried out with DAPI (Sigma, St. Louis, MO) for 1 minute. For negative controls, the primary antibody was omitted. Cells were analyzed under a fluorescent microscope (Nikon, Germany).
RNA extraction, cDNA synthesis, and reverse transcription-polymerase chain reaction
Total cellular RNA was extracted using Trizol reagent (Invitrogen, Grand Island, NY) according to the manufacturer's instructions and examined for purity and concentration using a photometer (Nanodrop ND). Possible DNA contamination was combated by DNase I (Sigma, St. Louis, MO) incubation for 30 minutes at 37°C. One microgram RNA and primers (Bioneer cyclescript RT premix) were used for cDNA synthesis (complementary cDNA synthesis kit; Bioneer, South Korea). Polymerase chain reaction (PCR) was performed using Gene Amp PCR system 9600 (PerkinElmer Life and Analytical Sciences, MA). Specific primer pairs of Oct4, Nanog, C-kit, Fragilis, Dazl, Mvh, Scp3, and Gdf9 (Table 1) were used for PCR. Cycling parameters were as follows: 5 minutes initial denaturation at 94°C, then 33 cycles at 94°C for 30 seconds, corresponding annealing temperature of primer pairs for 30 seconds, and 72°C for 1 minute. Ovarian cells from adult ovaries were used as positive control and liver cells were used as negative control. PCR products were loaded on 2% agarose gel, stained by ethidium bromide, and observed by UVI doc gel documentation system (Cambridge, CB4 1QB-UK). Expression of β-actin as a housekeeping gene was examined as internal control.
Isolation of granulosa cells from mouse ovarian tissue
To provide optimized condition and mimic natural environment of ovary for differentiation of OSCs into oocyte-like cells (OLCs), we designed a coculture system using OSC colonies from third passage associated with granulosa cells. According to previous reports and synchronization between granulosa cells and undifferentiated cells development, we used granulosa cells from 5 to 7 days mouse ovary (Qing et al., 2008).
For the isolation of granulosa cells from mouse ovary, O & Baker method was used (Wai-Sum and Baker, 1978). In brief, 5–7 days mouse ovaries were removed under dissecting microscope, and after washing with PBS and mincing into small fragments, samples were transferred into digestion solution (containing 1 mL 0.05% EDTA–Trypsin, 1 mL 0.2% collagenase IV, 50 μL 0.02% DNase I, and 2.5 mL DMEM). Enzymatic digestion was done within 30 minutes with continuous pipetting. The whole sample was then centrifuged at 300 g for 5 minutes after which the supernatant was discarded, and the pellet was resuspended by adding fresh prewarm DMEM. Finally, cells were transferred into a tissue culture flask and kept for one night in 37°C. The next day, cells were washed with PBS three times, and after adding culture medium, they were kept for 2 weeks. To employ granulosa cells as a feeder layer in a coculture system, there was the need to inactivate the cells. The granulosa cells were thus treated with Mitomycin-C (10 μg/mL, Sigma) for 2 hours.
Meanwhile, to check for a potential contamination with germ cells and pluripotency markers during the granulosa cell isolation procedure, after 2 weeks of culture, molecular evaluation was done for genes related to oocyte such as Gdf9, Fig α, Zp2, and Mvh, and genes related to pluripotency and stem cell such as Oct4, Nanog, C-kit, Fragilis, and Dazl by reverse transcription-polymerase chain reaction (RT-PCR).
OSCs and granulosa cells coculture
Mitomycin-C-treated granulosa cells were transferred onto gelatin-coated four-well plates (5 × 104/cm2) and incubated for one night at 37°C. OSC colonies were then transferred to granulosa cell culture plates with a sampler. We noticed that OSC colonies were highly compact and dense and colony disassociation with routine enzymatic and mechanical digestion was impossible; therefore, we harvested each colony with capillary pipette and transferred each colony by sampler onto granulosa cells feeder layer. This coculture system was maintained for 11 days while changing the medium every 2 days.
The culture media that we used for differentiation purpose were those that we used for OSCs propagation without LIF.
Immunofluorescence evaluation of differentiated OLCs
After 11 days in the coculture, OLCs were subjected to immunostaining for the presence of synaptonemal complex 3 (SCP3) and growth differentiation factor 9 (GDF9) proteins. The procedures used for this section were as same as those we used for immunostaining before differentiation. SCP3 is a meiosis-specific protein the presence of which in OLCs indicates entry into meiosis, whereas GDF9 is an oocyte-specific protein. The antibodies we used in our research include rabbit polyclonal antibody to SCP3 (ab15093; Abcam) and goat polyclonal secondary antibody to rabbit IgG (ab6717; Abcam). For SCP3 protein localization in OLCs and examination of GDF9 protein in OLCs, we used goat polyclonal IgG to GDF9 (sc-12244; Santa Cruz Biotechnology) and donkey polyclonal secondary antibody to goat IgG FITC (ab6881; Abcam).
Molecular and morphological examination of differentiated OLCs
In addition to immunofluorescence evaluations, we performed a molecular and morphological evaluation for differentiated OLCs. Molecular evaluation was done on Scp3 and Gdf9 genes by RT-PCR, and after trypsinization of differentiating colonies, cells were observed under light microscope and their sizes and shapes were analyzed by ImageJ software. All experiments were repeated independently at least three times.
Results
Isolation of OSCs by preplating technique
For the isolation of stem cells from neonatal ovary and establishment of OSC line in vitro, we applied the protocol used for male germ stem cells (GSCs) with slight modifications. After the enzymatic digestion of ovarian tissue, preplating technique was used for the elimination of possible fibroblast and somatic cells contamination. This technique is based on the differential adherence characteristics of primary culture cells to a gelatin-coated surface. After digestion, whole cell suspension was transferred to a gelatin-coated dish and primary culture was done at 37°C for 30 minutes. After the 30 minutes, buoyant cells were collected, and after adding culture medium, they were transferred to a culture dish (secondary culture).
During the first 4 days, colonies were formed with undefined border (Fig. 1a, b). Cell passaging during first 3–4 days accelerated cell proliferation and colony formation. During 8–10 days, the size of colonies increased rapidly and colony appearance became similar to those seen in embryonic stem cell colonies (Fig. 1c–e). Colonies formed by the preplating technique were highly compact and dense, making cell disassociation by adding trypsin as well as cell counting impossible.
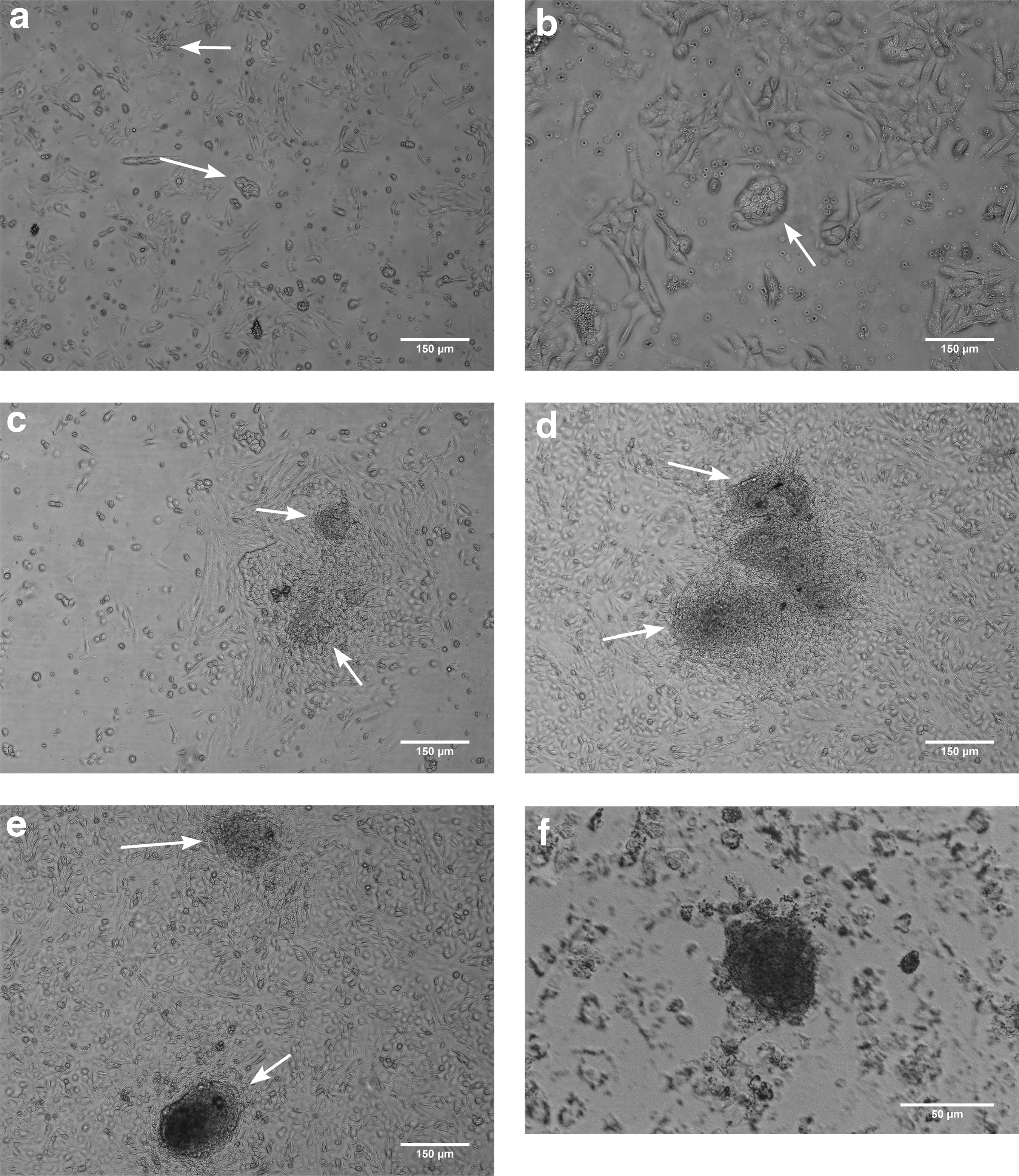
Morphological features of colony-forming OSCs.
Immunofluorescence analysis and alkaline phosphatase activity
After morphological examinations, colonies were evaluated for alkaline phosphatase activity as well as presence of OCT4, SSEA1, DAZL, and MVH proteins.
Our data showed that these colonies had alkaline phosphatase activity as a germ cell marker (Fig. 1f). Also, immunofluorescence analysis confirmed the presence of OCT4, SSEA1, Dazl, and Mvh positive cells in our colonies. Owing to high density of the colonies, cells located in the center of the colonies were unclear (Fig. 2).

Immunofluorescence of colonies against stem and germ cell-specific markers. OCT4
RT-PCR results
RT-PCR analysis was performed to evaluate the expression of Oct4 (stem cell and germ cell marker), C-kit (stem cell marker), Nanog (pluripotent marker), Fragilis (germ cell-specific marker), Dazl and Mvh (specific to germ cells), Scp3 (meiosis-specific marker), and Gdf9 (oocyte-specific marker) genes in the colonies. Our data showed that the cells highly expressed genes related to pluripotency and germ-line stem cell markers (Fig. 3); however, they did not express differentiated germ cells and oocyte-like markers such as Scp3 and Gdf9 (data not shown). Mouse ovary was used as positive control whereas β-actin was considered as internal control, and all experiments were independently repeated three times.
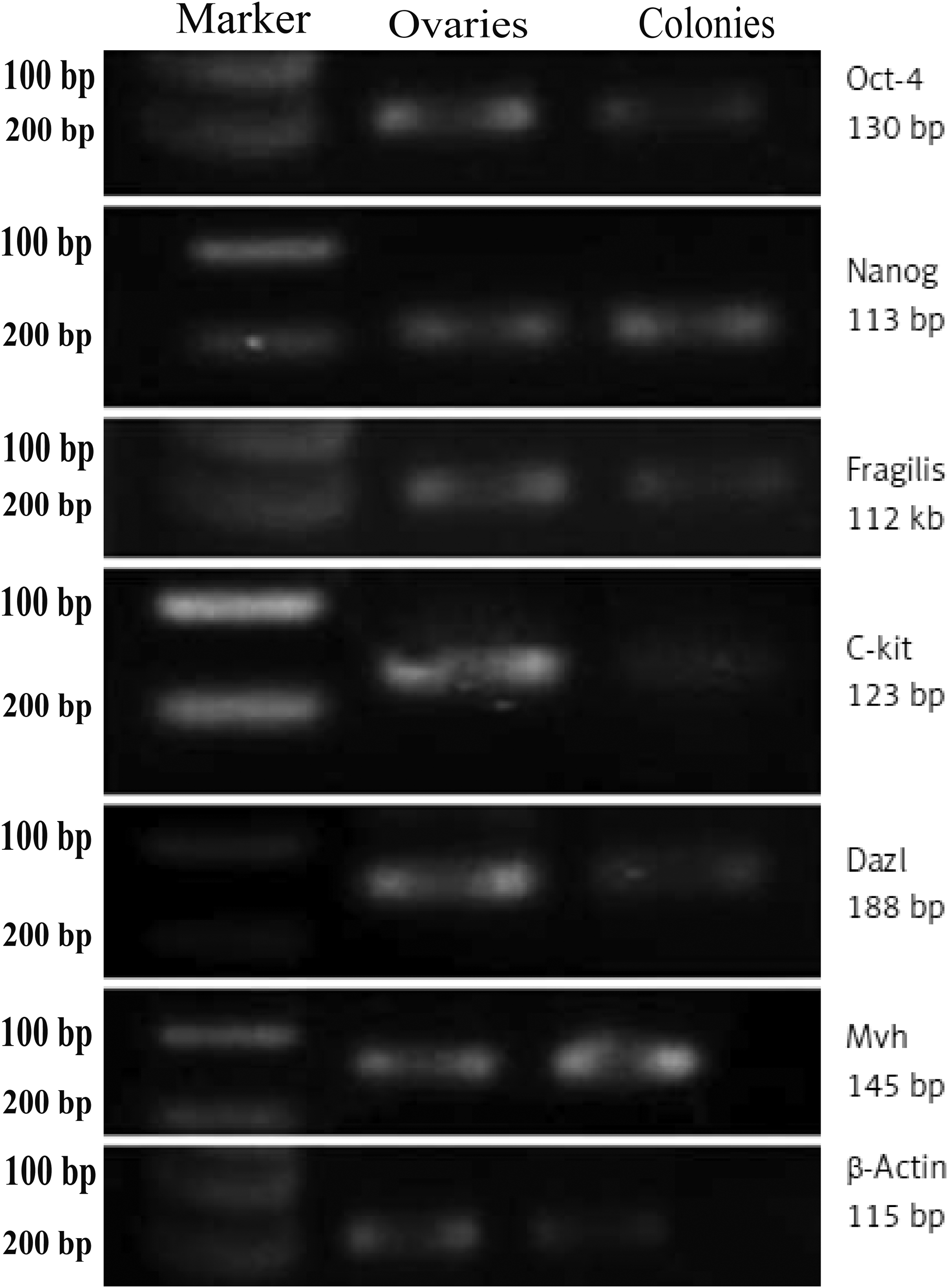
The RT-PCR analysis of stem and germ cell-specific markers in colony-forming cells. Bands related to the expression of Oct4, Fragilis, C-kit, Nanog, Mvh, and Dazl were observed on gel in the right predicted size. Cells did not express Scp3 and Gdf9 (data not shown). β-Actin as a housekeeping gene was also studied. Expression profile of ovarian cells from adult ovaries and liver cells was used as positive and negative controls, respectively (Parvari et al., 2015).
Isolation of granulosa cells and cultivation
The obtained granulosa cells through the O & Baker protocol were cultured. These cells like other somatic cells had fusiform appearance and could grow finely to reach maximum confluency in culture dishes in 7–11 days (Fig. 4a).
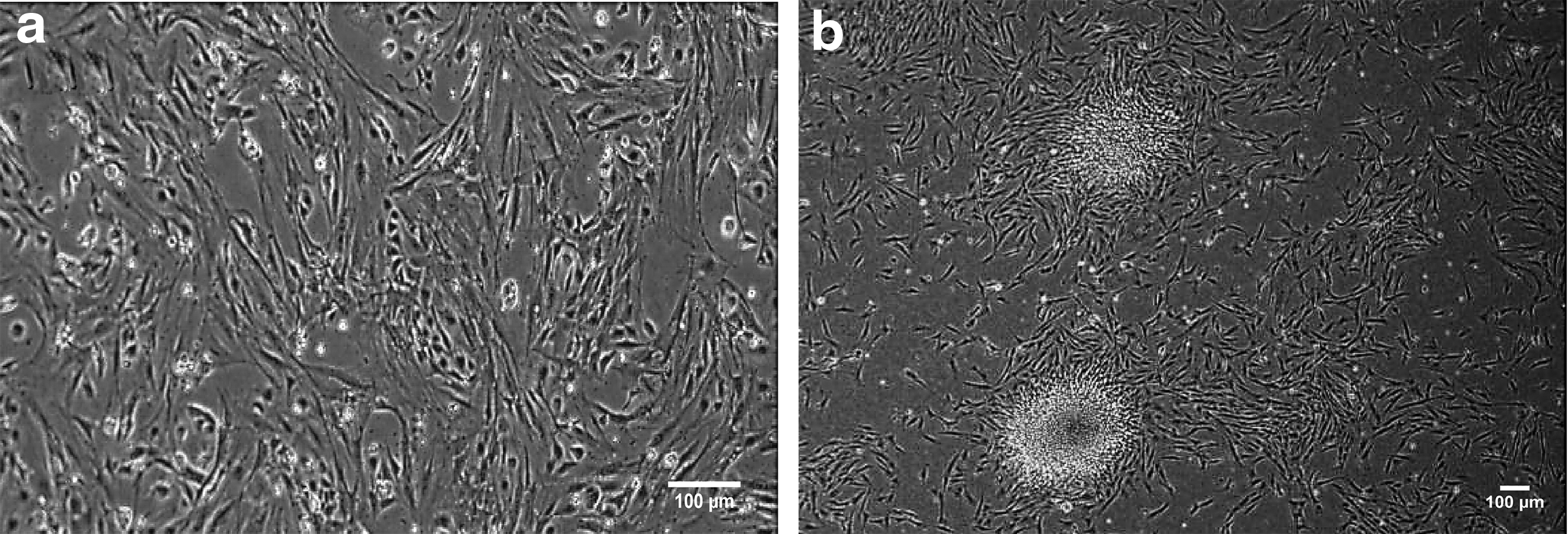
Cultivation of mouse granulosa cell and coculture with OSC colonies.
Molecular evaluation of granulosa cells
Despite the established and accepted O & Baker protocol for isolation of granulosa cells, to confirm that no contamination of isolated cells with germ cells and other stem cells occurred, we performed evaluation of oocyte-specific genes by RT-PCR for Gdf9, Mvh, Oct4, Nanog, C-kit, Fragilis, and Dazl genes after 2 weeks of culture, and ultimately our data showed that none of these genes were expressed in our isolated cells (Fig. 5).
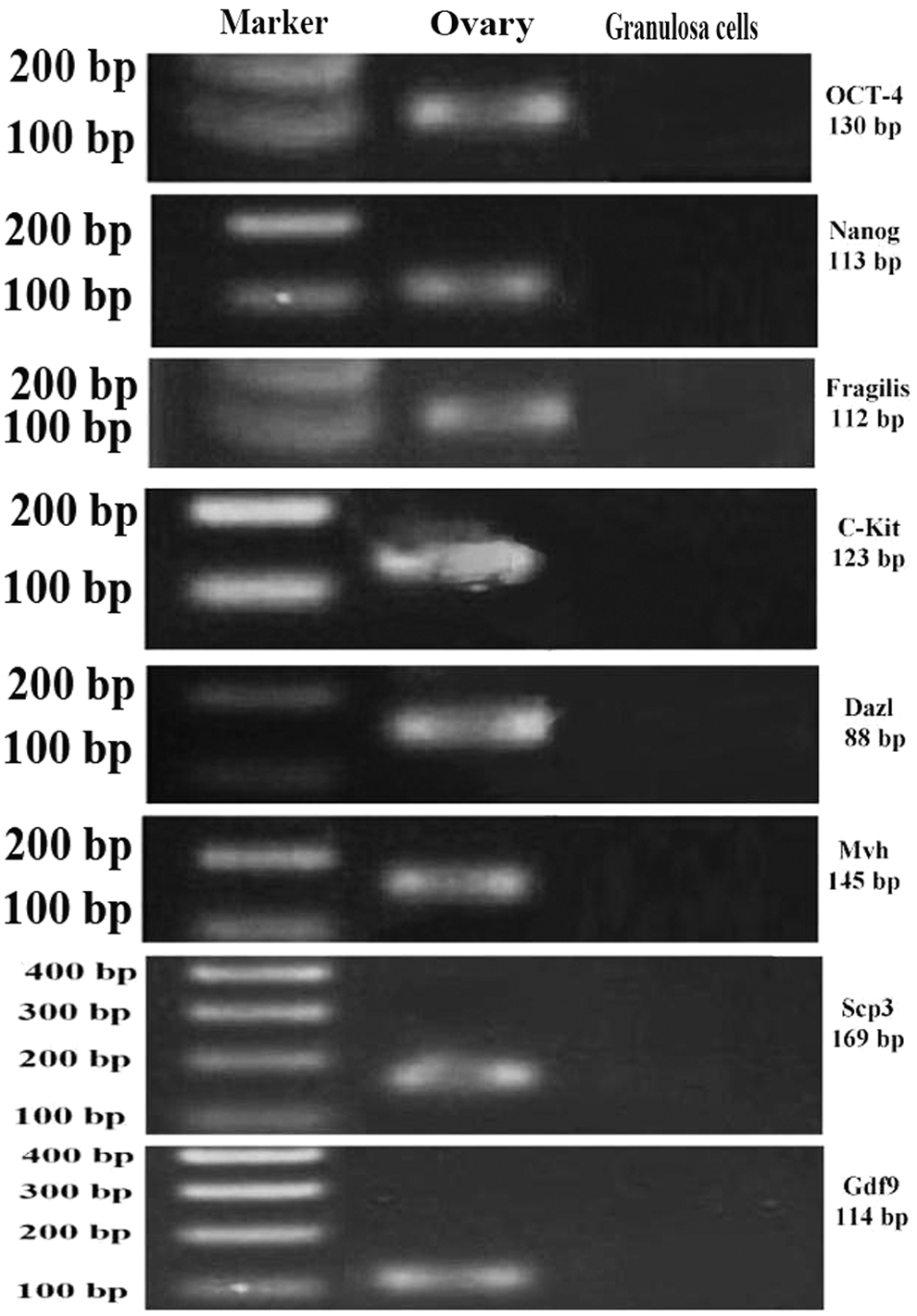
The RT-PCR analysis of stem and germ cell-specific markers in granulosa cells after 2 weeks of culture. Isolated granulosa cells did not express oocyte, pluripotency, and stem cell-related genes after 2 weeks of culture after isolation. Ovarian cells from adult ovaries were used as positive control.
OSCs colony transfer to granulosa cells feeder layer for differentiation induction
The granulosa cells, after treating with mitomycin-C (10 μg/mL) and inactivated, were transferred onto gelatin-coated four-well plates (5 × 104/cm2) and incubated for one night at 37°C. On the next day, the medium was discarded, and stem cell colonies and differentiation medium were added onto granulosa feeder layer (Fig. 4b).
Evaluation of differentiated OLCs
After 11 days in the coculture, cells were subjected to different evaluations:
Immunofluorescence evaluations
After passing the needed time for differentiation, cells were assessed for oocyte-specific proteins such as SCP3 and GDF9 (Fig. 6).
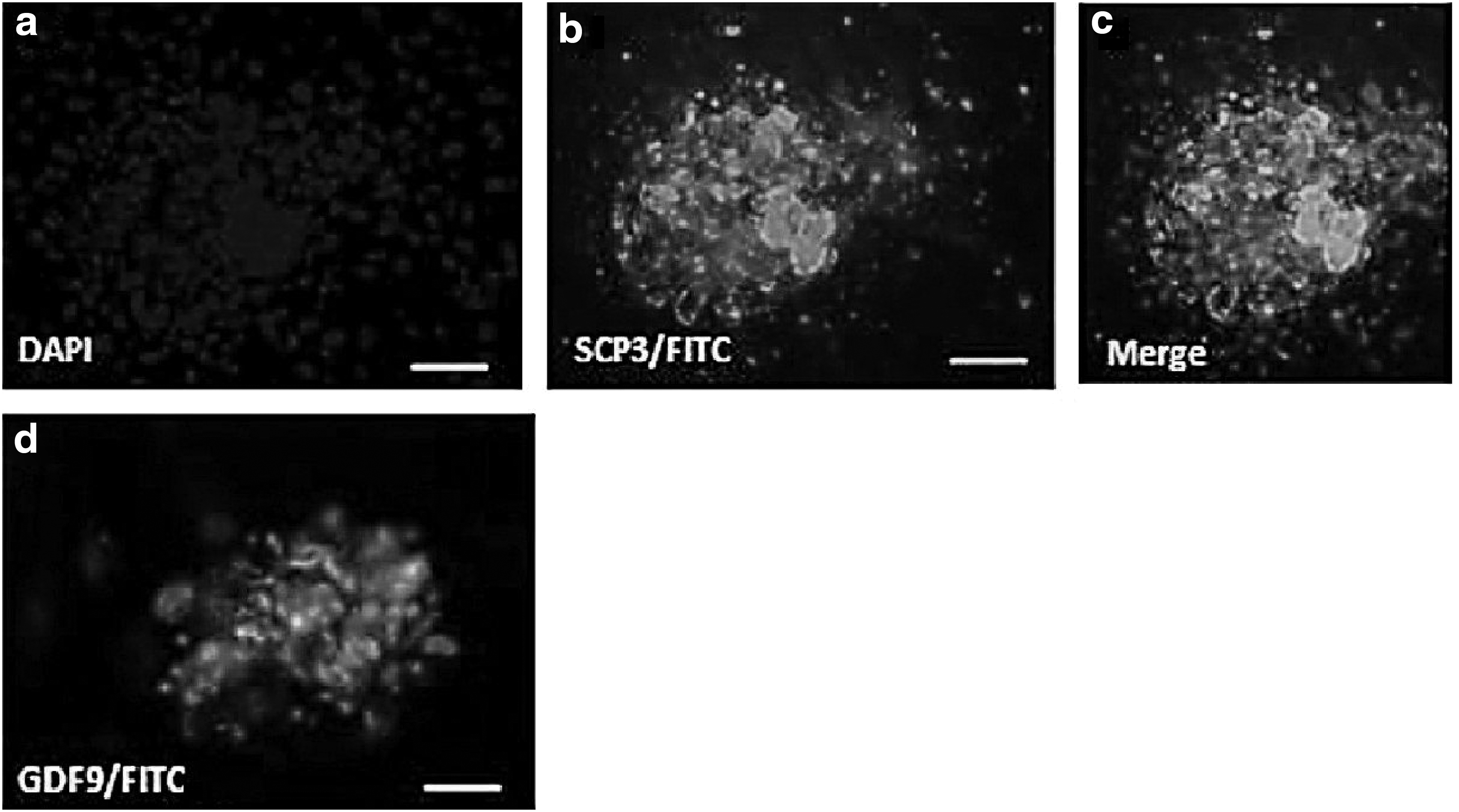
Protein expression of SCP3 and GDF9 in differentiating colonies (
Molecular evaluations
Cellular assessment using the RT-PCR technique showed that Scp3 and Gdf9 genes have been expressed (Fig. 7).

Expression of oocyte-specific genes in differentiated colonies. Scp3 and Gdf9 genes were expressed in differentiated samples, but not in proliferation.
Morphological evaluations
After 11 days, cells gradually started to propagate, but this propagation was not significant and the overall appearance was not changed considerably. After trypsinization, cells were observed under microscope and it was clear that the size of some cells had increased and reached 30–40 μm (Fig. 8).

Differentiated cells morphology. Cellular size has increased in some cells. Arrows show size-increased cells.
Discussion
In this study, our aim was to evaluate differentiation induction of OSCs by the coculture method with granulosa cells, and ultimately we found that granulosa cells have great potential for this purpose. Results from this study indicated that by the preplating technique, isolation and colony formation of OSCs are much easier and more affordable than other techniques such as FACS and MACS. Moreover, alkaline phosphatase activity, immunofluorescence, and RT-PCR results confirmed our isolated germ line stem cell identity. In addition, the coculture system showed that the presence of granulosa cells is vital for the accurate differentiation of GSCs by direct contact with germ cells during oocyte development, and this may have resulted from the absence of cellular transport system in oocytes. In some researches, it has been elucidated that receptor–ligand systems such as Kit and Kit ligand that exist between granulosa and oocyte cell are one of the most important regulation systems during early follicular growth, which by antiapoptotic function cause better growth and differentiation of germ cells in gonads (Kerr et al., 2008).
Despite denial of the presence of OSCs in postnatal ovaries by some researchers, our results show that OSCs are not only present in postnatal ovary but also they have differentiation potential into oocyte, and besides new progress in stem cell therapy, these cells are a new promising treatment for women who suffer from infertility because of DOR, POI, and POF disorders.
The presence of stem cells in a variety of adult tissues such as bone, brain, dental pulp, skin, adipose tissues, and testis has been proved for many years (Daar et al., 2004), but there is no consensus among scientists regarding the presence of stem cells in postnatal ovaries. In fact, it has been accepted that all germ cells of female mammals are generated from precursor cells called PGCs and oogenesis is completed and ceased before birth (Zuckerman, 1951). Therefore, female neonatal mammals are endowed at birth with fixed number of germ cells (oocytes) enclosed within follicles, and after birth, renewal capacity does not exist anymore and the number of germ cells reduces with advancing age (Di Carlo et al., 2000). The hypothesis that there is no neo-oogenesis after birth in mammals has been challenged recently by Tilly group (Johnson et al., 2004, 2005; White et al., 2012; Woods and Tilly, 2013). They did not only claim that juvenile and adult mouse ovaries possess mitotically active germ cells, but they could also introduce a comprehensive method for isolation of these stem cells from mouse and human ovary by the DDX4 marker (White et al., 2012; Woods and Tilly, 2013). In addition, Virant-Klun et al. applied a simple procedure for isolation of female germ line stem cells by scraping of the OSE. Although they were able to isolate and characterize these cells, this method was inapplicable in smaller mammals such as mouse (Virant-Klun et al., 2008, 2009, 2011). Moreover, Zou et al. (2009, 2011) could isolate and propagate female germ line stem cells from mouse ovary by MVH and Fragilis surface markers by the MACS technique. There is another strategy for this purpose that has been used by Pacchiarotti et al. (2010) who could isolate germ line stem cells from ovarian tissue by using transgenic mice, but this strategy is so complicated that makes the interpretation of obtained results difficult.
Guan et al. (2009) published and described different protocols used for isolation of spermatogonial stem cells (SSCs) from testis. In that article, they introduced a novel method for isolation of SSCs, which after enzymatic digestion, the obtained cells were transferred to culture medium. After colony formation, those colonies that have appearance same as SSC colonies were selected. They called this method the morphology-based selection method. Also, Gong et al. (2010) cultivated obtained cells from enzymatic digestion of ovarian tissue and reported that the colonies formed from this method are similar to those colonies resulted from mouse embryonic stem cells. In this study, we used the morphology-based selection method for our isolation procedure because this method is simple and easy to perform and is more affordable than the other methods.
Two groups have independently reported that OSCs obtained from mouse and human ovaries differentiate into OLCs spontaneously without any somatic feeder cell, and that these differentiated OLCs have large spherical appearance and gene expression profile same as oocyte (Pacchiarotti et al., 2010; White et al., 2012). Although in vitro-derived from morphological and gene expression profile aspect are similar to in vivo-derived oocytes, these cells clearly have not sufficient competency for fertilization or embryonic development. Probably, presence of the somatic companion of germ cells in ovary, granulosa cells, is vital and essential for accurate differentiation of OSCs, and is required for periodic meiotic arrest and proper imprinting.
Pacchiarotti et al. (2010) showed that aggregation of the GSC line derived from neonatal mouse ovary with dispersed ovaries of neonatal mice led to follicle-like structure formation, and gene expression profile of the differentiated oocyte-like structures showed that they expressed oocyte-specific markers, including Ybx2, Gdf9, and Zp1 at early stages during embryonic body formation. Also, there are some other reports indicating that aggregation and removal of growth factors from mouse embryonic stem cell cultures induce an efficient oocyte differentiation (Hübner et al., 2003). Moreover, in a single study that was done by Park et al. (2013), it was demonstrated that by using specific growth factor bone morphogenetic protein 4, differentiation of OSCs into oocyte-like structures increased in comparison with Noggin-treated groups. Meanwhile, other studies indicated that injection of GFP-positive OSCs into adult ovarian tissue of mouse or human will produce fertilization-competent eggs and viable embryos and offspring (White et al., 2012; Zou et al., 2009).
In our study, formed colonies consisted of round and small cells, which beside their pluripotency properties were similar to very small embryonic-like cells (VSELs) (Kucia et al., 2006). In a similar study, Honda et al. (2007) observed round colonies after ovarian enzymatic digestion and cultivation of obtained cells, which, like embryonic stem cells colonies, were too compact with small cells and alkaline phosphatase activity. Recently, Bhartiya et al. (2015) showed that adult human ovaries possess two kinds of cellular population, VSELs and slightly larger progenitors equivalent to SSCs in the testes, which have the ability to undergo neo-oogenesis in adult ovaries, differentiate into oocytes, and undergo primary follicle assembly under the influence of FSH.
In immunofluorescence evaluations after differentiation, our finding showed that SCP3 and GDF9 were expressed in the margin and special location in the colonies. This pattern of OSCs differentiation in in vitro condition is comparable with the pattern of differentiation of PGCs from epiblast during mouse early embryo development such that germ cells only originate from the most posterior of the proximal part of the epiblast. It seems that the distal part of the epiblast lacks intracellular signals such as Smads signaling pathway, and the cells present in the margin of the colonies may possess intracellular signaling of Smads pathway for differentiation (Hayashi et al., 2002). Recently, Parte et al. have evaluated the spontaneous differentiation of OSCs in vitro. Parte et al. (2014) indicated that after 1 week of culture, not only did the OSCs became bigger in size and gained abundant cytoplasm and balbiani-like structure but also exhibited cytoplasmic streaming and gene expression pattern as those seen in oocyte (Dazl, Gdf9 and Scp3).
In this study, after differentiation, we performed cell separation using trypsin after which we did microscopic evaluations. The microscopic evaluations showed that although we had an increase in cell size, structures such as oocyte or follicle were not seen. These results are comparable with the results of Qing et al. (2007), so maybe these cells were in their early development stage.
In summary, our results in this study show that although the presence of OSCs in adult ovaries is still denied by some researchers, their existence is an undeniable fact and not only do they exist but also they have oogenic potency either by induction or by spontaneity if their microenvironment and required growth factors are provided. Presence of OSCs in adult human ovaries is a promising fact, which, by further investigations in the field of stem cell therapy, could be used either for autologous treatment of ovarian infertility in the future or as an in vitro model to study the development of mammalian oocytes. However, to achieve these aims, more investigations and researches are needed.
Footnotes
Acknowledgment
This article is part of the thesis for a PhD that was supported by Grant 89 01-30-10462 from Tehran University of Medical Sciences.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.