
Abstract
Abstract
In our previous study, we found that treatment of miniature pig somatic cell nuclear transfer (SCNT) embryos with 4 mM valproic acid (VPA), a histone deacetylase inhibitor, for 48 hours after activation enhanced blastocyst formation rate and octamer-binding transcription factor-3/4 (Oct-3/4) gene expression at the late blastocyst stage; however, the production of viable cloned pups failed, when those VPA-treated SCNT embryos were transferred to recipients. This failure suggests that the present VPA treatment is suboptimal. In the present study, we explored the optimal conditions for VPA to have beneficial effects on the development of SCNT embryos. When miniature pig SCNT embryos were treated with 8 mM VPA for 24 hours after activation, both the rates of blastocyst formation and blastocysts expressing the Oct-3/4 gene were significantly (p < 0.05) improved. A similar increase in blastocyst formation was also observed when microminipig-derived cells were used as SCNT donors. Five cloned piglets were obtained after the transfer of 152 microminipig SCNT embryos that had been treated with 8 mM VPA for 24 hours. The results indicated that a short duration of treatment with VPA improves the development of both miniature pig and microminipig SCNT embryos, possibly via an enhanced reprogramming mechanism.
Introduction
P
Unfortunately, the cloning efficiency of these pigs is very low, and this has been an obstacle to their use in various biomedical applications. After SCNT, gene expression in the transferred somatic cell nuclei becomes reprogrammed to mimic that of preimplantation embryos (De Sousa et al., 1999). However, Niemann et al. (2002) demonstrated that gene expression patterns in SCNT embryos differed from those in in vitro-fertilized embryos, suggesting that the low efficiency in SCNT may be due to incomplete reprogramming in the transferred donor nuclei.
Therefore, development of a method to monitor the levels of reprogramming in donor nuclei would be necessary to predict the success of in vivo development of SCNT embryos. In a previous study, we developed a simple, noninvasive monitoring system for evaluation of the status of the octamer-binding transcription factor-3/4 (Oct-3/4) promoter gene in miniature pig SCNT embryos during in vitro development, using the Oct-3/4 promoter-driven enhanced green fluorescent protein (EGFP) (Miyoshi et al., 2009). Oct-3/4, also known as POU5F1 (POU domain, class 5, transcription factor 1), is a protein that is specifically expressed in the primordial germ cells, ovary, and testis, but not in the heart, liver, lung, kidney, spleen, and fibroblasts (Lee et al., 2006). Its expression has also been reported in pig preimplantation embryos (Kirchhof et al., 2000; Vejlsted et al., 2006). Therefore, Oct-3/4 is believed to be closely related to nuclear reprogramming activities in SCNT embryos. Based on these observations, our Oct-3/4 promoter-EGFP-based system would be useful to evaluate the levels of reprogramming in SCNT embryos.
Valproic acid (VPA), a cell-permeable, short-chain fatty acid that inhibits histone deacetylase, is known as a useful drug that induces the reprogramming of differentiated cells (Huangfu et al., 2008a, 2008b). We previously examined whether VPA is beneficial to the in vitro development of miniature pig SCNT embryos, and it is capable of upregulating endogenous Oct-3/4 gene expression by using the Oct-3/4 promoter-EGFP-based system mentioned earlier. As a result, we found that treatment with 4 mM VPA for 48 hours after activation enhances the ability of miniature pig SCNT embryos to develop into blastocysts and maintains their ability to express the Oct-3/4 gene (Miyoshi et al., 2010a). This means that VPA is able to improve cloning efficiency. Therefore, we transferred these VPA-treated SCNT embryos to recipient females; however, this attempt failed, as no cloned miniature pigs were obtained (Miyoshi et al., unpublished data). This failure could be attributed to the extended duration of the in vitro culture of SCNT embryos before transfer to the recipients, because the in vitro culture conditions of pig embryos are still considered suboptimal (Grupen, 2014). Prolonged exposure of embryos to VPA could also be one of the critical factors leading to pregnancy failure.
In the present study, we explored VPA treatment conditions that facilitate favorable reprogramming induction and in vitro development of miniature pig SCNT embryos. In addition, we tested whether it is possible to produce viable cloned microminipigs derived from SCNT embryos, using the optimal VPA treatment conditions that we developed.
Materials and Methods
Experiments with recombinant DNA technology were performed in agreement with the guidelines of Kagoshima University Committee on recombinant DNA security. All animal experiments in the present study were approved by the Animal Ethics Committee, Kagoshima University.
Construction of plasmids
Plasmid pdOEN-2 (Fig. 1) was constructed by placing a proximal portion of mouse Oct-3/4 promoter in pGOF-18 (Yeom et al., 1991) upstream of EGFP cDNA+poly(A) sites of the SV40 gene in pEGFP-N1 (Clontech, Palo Alto, CA), from which the cytomegalovirus (CMV) promoter had been deleted. First, pEGFP-N1 was digested with AseI and NheI, blunted with T4 DNA polymerase (Toyobo, Tokyo, Japan), and finally self-ligated to create CMV promoter-free pdEGFP-N1. The ∼3-kb fragment containing the proximal portion of mouse Oct-3/4 promoter formed through digestion of pGOF-18 with BglII and SalI was then inserted into the BglII and SalI sites of pdEGFP-N1 to create pdOEN-2. Fidelity of the junctional portion spanning the Oct-3/4 promoter and EGFP cDNA was confirmed by sequencing. For linearization, pdOEN-2 was digested with BglII. The plasmid pAQC (Sato et al., 2012b) (Fig. 1) was designed to express 2 transgenes: hygromycin B resistance gene under control of the mouse phosphoglycerate kinase promoter, and tandem dimer Tomato (tdTomato) cDNA under control of the chicken β-actin-based CAG promoter (Niwa et al., 1991). The two units were aligned in a head-to-head fashion. For linearization, pAQC was digested with HindIII. These plasmids were amplified in DH5α Escherichia coli, propagated on an agar plate, and purified by using a method described by Sato et al. (2012a).
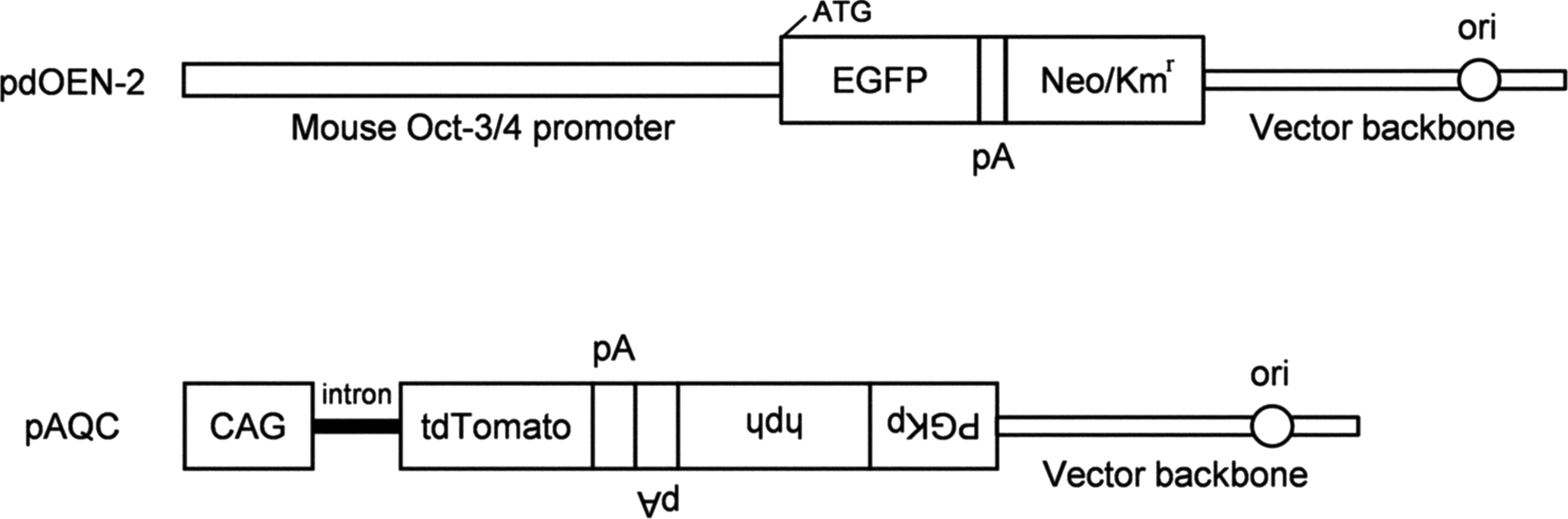
pdOEN-2 and pAQC plasmids used for transfection of CKCs. CAG, cytomegalovirus enhancer+chicken β-actin promoter; CKCs, Crawn miniature pig kidney-derived cells; EGFP, enhanced green fluorescent protein cDNA; hph, hygromycin B resistance gene; Kmr, kanamycin resistance gene; Neo, neomycin resistant gene; ori, replication origin; pA, poly(A) sites; PGKp, mouse phosphoglycerate kinase promoter; tdTomato, tandem dimer Tomato cDNA.
Donor cells
Linearized pdOEN-2 and pAQC plasmids were transfected into Crawn miniature pig kidney-derived cells (CKCs) (Ozawa et al., 2015), using a nucleofector-mediated transfection system (Lonza Biologics, Cologne, Germany) and the modified methods of Nakayama et al. (2007). For transfection, a 100 μL solution containing 10 μL of plasmid DNA (6 μg) and 90 μL of Opti-MEM (Invitrogen Co., Carlsbad, CA) was mixed with ∼0.5 × 106 cells, and the mixture was then placed into an Amaxa cuvette with a 2.5 mm gap. Cells were then nucleofected by using a pre-programmed setting (V010) on the Amaxa nucleofector apparatus. After electroporation, the cells were split into three 60-mm gelatin-coated dishes (Iwaki Glass, Tokyo, Japan), and they were further incubated in medium containing 400 μg/mL of G418 (Invitrogen Co.) and 40 μg/mL of hygromycin B (Invitrogen Co.) for an additional 10–15 days. Colonies (containing 300–700 cells) were selected by using a small paper disk (3MM Whatman paper, 5 mm in width, and 5 mm in length) that had been dipped in 0.125% (w/v) trypsin/0.01% (w/v) EDTA.
Cells were directly transferred to wells of a gelatin-coated 48-well plate (Iwaki Glass) that contained hygromycin B/G418 medium. The medium used was a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium (DMEM/F12; Wako Pure Chemical Co., Osaka, Japan), which was supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen Co.) and 1% (v/v) antibiotic-antimycotic solution (Sigma-Aldrich Co. Ltd., St. Louis, MO). The cells were cultured for 20–30 days until they reached confluency at 37°C in a humidified atmosphere of 5% CO2 in air. These cells were further propagated in a stepwise manner, checked for the uniform expression of tdTomato-derived fluorescence (but not EGFP-derived fluorescence) under ultraviolet (UV) light, and finally frozen. A portion of each clone was subjected to polymerase chain reaction (PCR) analysis for confirmation of the presence of pdOEN-2 and pAQC transgenes in its genome (data not shown). One of these clones was used as the donor for SCNT.
In addition, the kidneys of an 18-month-old female and a 3-year-old male microminipig were resected. Ear skin biopsies were obtained from two newborn female and three newborn male microminipigs, and four microminipig fetuses were obtained on day 30 of pregnancy. The renal cortex tissues, skin biopsies, and fetal body tissues were cut into small pieces, transferred into individual 50-mL tubes, and enzymatically digested with 0.25% trypsin-EDTA (Gibco BRL, Grand Island, NY) for 15 minutes at 37°C, while being stirred. After an equal volume of FBS was added to each tube, the digested tissue was allowed to settle for 5 minutes, and the supernatant containing the disaggregated cells was centrifuged at 190 g for 5 minutes. The cell pellet was resuspended and cultured in DMEM/F12 that was supplemented with 100 μg/mL amikacin sulfate and 10% FBS, under 5% CO2 in air at 37°C. Cells were passaged after reaching confluence and used as donors for SCNT at passages 5 and 10 of culture.
The cells were allowed to grow to confluency and cultured for an additional 5–6 days without a change of medium. A single-cell suspension was prepared by standard trypsinization immediately before SCNT.
In vitro maturation of oocytes
Ovaries were collected from prepubertal gilts at a local slaughterhouse and transported to the laboratory in saline at 32°C–35°C. The follicular contents were recovered by aspiration from follicles (2–5 mm in diameter), by using an 18-gauge needle (Terumo Co., Tokyo, Japan) and a 5-mL disposable syringe (Nipro Co., Osaka, Japan). The cumulus-oocyte complexes (COCs) were gathered from the follicular contents and washed twice with HEPES (Nacalai Tesque Co., Kyoto, Japan)-buffered Tyrode-lactate-pyruvate-polyvinyl alcohol (PVA; Sigma-Aldrich Co. Ltd.) and the maturation medium, respectively. The maturation medium consisted of 90% (v/v) TCM-199 with Earle's salts (Gibco BRL), supplemented with 0.91 mM sodium pyruvate (Sigma-Aldrich Co. Ltd.), 3.05 mM D-glucose (Wako Pure Chemicals Co.), 0.57 mM cysteine hydrochloride hydrate (Sigma-Aldrich Co. Ltd.), 10 ng/mL epidermal growth factor (Sigma-Aldrich Co. Ltd.), 10 IU/mL equine chorionic gonadotropin (eCG; ASKA Pharmaceutical Co., Tokyo, Japan), 10 IU/mL human chorionic gonadotropin (hCG; ASKA Pharmaceutical Co.), 100 μg/mL amikacin sulfate (Meiji Seika, Tokyo, Japan), 0.1% (w/v) PVA, and 10% (v/v) pig follicular fluid. Only COCs possessing a compact cumulus mass and evenly granulated ooplasm were selected.
COCs in groups of 30–50 were transferred to a droplet of the maturation medium (200 μL) under paraffin oil (Nacalai Tesque Co.) in a 35-mm dish (Becton Dickinson, Fullerton, CA) and were cultured at 38.5°C in an atmosphere of 5% CO2 in air. During culture, mechanical vibration at a frequency of 20 Hz and accelerations of ±0.33 G and ±0.11 G in the x- and y-axis directions, respectively, were applied to the COCs for 10 seconds at intervals of 60 minutes by using an NSSB-200 mechanical vibration instrument (Nepa Gene Co., Chiba, Japan) (Mizobe et al., 2010). After 40–42 hours of culture, cumulus cells were removed by pipetting with 0.1% (w/v) hyaluronidase (Sigma-Aldrich Co. Ltd.). Oocytes with a polar body were selected for further investigation.
Production of SCNT embryos
In vitro-matured and denuded oocytes were transferred into HEPES-buffered TCM-199, with the osmolarity adjusted to 300 mOsm by the addition of sucrose supplemented with 5 μg/mL cytochalasin B and 10% FBS. The oocytes were enucleated by removal of the first polar body and the metaphase II plate in a small amount (∼20% of total volume) of surrounding cytoplasm, with a 15-μm inner diameter glass pipette. A single donor cell was inserted into the perivitelline space of each enucleated oocyte by using the same glass pipette. Cell-oocyte complexes were transferred to modified porcine zygote medium-3 (mPZM-3) (Sato et al., 2005) and were kept at 38.5°C, in 5% CO2 in air until fusion. The chamber for fusion was a 60-mm dish (Becton Dickinson) filled with 7 mL of fusion medium, composed of 250.3 mM sorbitol, 0.5 mM Mg(CH3COO)2, 0.5 mM HEPES, and 0.2% (w/v) bovine serum albumin (BSA).
Two stainless-steel wires (100 μm in diameter) were used as electrodes that were attached to micromanipulators. The single cell-oocyte complex was sandwiched between the electrodes and was oriented with the contact surface between the cytoplast and the donor cell, perpendicular to the electrodes. Membrane fusion was induced by applying a single direct-current pulse of 25 V for a duration of 20 microseconds with a prepulse of alternating-current field of 5 V, 1 MHz for 2 seconds using an LF 101 Fusion Machine (Nepa Gene Co.). After the fusion pulse, the complexes were cultured for 2 hours in 100 μL of mPZM-3 until activation. Fusion was determined by microscopic examination, 1 hour after the pulse was applied.
Activation and culture of SCNT embryos
Fused embryos were activated by ultrasound (for embryos derived from intact or transfected CKCs) or electric (for embryos derived from microminipig cells) stimulation. For ultrasound activation (Miyoshi et al., 2007), embryos were washed twice in activation medium composed of 250.3 mM sorbitol, 0.1 mM Ca(CH3COO)2, 0.5 mM Mg(CH3COO)2, and 0.1% BSA, and they were then transferred to a well of a four-well plate (Nunc, Roskilde, Denmark) containing 700 μL of the medium. The ultrasound probe (8 mm in diameter) of a KTAC-3000 Sonopore (Nepa Gene Co.) was inserted directly into the activation medium, and the embryos were exposed to 2068 kHz at an intensity of 65 V for 30 seconds with a 10 Hz burst rate and a 10% duty cycle. A miniature stirrer was placed within the well and spun at 300 rpm during ultrasound exposure. For electric activation, embryos were washed twice in the activation medium, and they were then placed between two wire electrodes (1 mm apart) of the chamber slide in 15 mL of the medium. A single direct-current pulse of 100 V/mm was applied to the embryos for a duration of 50 microseconds by using an LF 101 Fusion Machine.
After each treatment, the embryos were cultured in 50 μL of mPZM-3 supplemented with different concentrations of VPA, under 5% CO2, 5% O2, and 90% N2 at 38.5°C. During the first 2 hours of culture, 2.2 μg/mL cytochalasin B was added to the medium to prevent extrusion of a polar body-like structure. After 24 hours of culture, the embryos were then transferred to 50 μL of mPZM-3 for further culture. The embryos were assessed for cleavage and blastocyst formation at 2 and 7 days of culture, respectively.
At the end of the culture, blastocysts were placed on slides with a drop of mounting medium, consisting of glycerol and phosphate-buffered saline (9:1) containing 100 μg/mL Hoechst 33342 (Sigma-Aldrich Co. Ltd.). A cover slip was placed on top of the blastocysts, and the edge was sealed with nail polish. The number of nuclei was counted under UV light. Some embryos were cultured for 7 days, and the resultant blastocysts were assessed for the expression of tdTomato under a fluorescence microscope. The blastocysts expressing tdTomato were placed on slides with the mounting medium, covered with a cover slip, and examined for the expression of EGFP under UV light (because it was faint).
Transfer of embryos
Two microminipig gilts, aged 20 and 21 months, were used as recipients. Estrus detection in these animals had been defined as previously described (Noguchi et al., 2015). At least two estrous cycles of normal length (∼20 days) and normal duration of estrus (∼3 days) were observed in each gilt before the start of the experiment. The recipients were given three intramuscular injections of estradiol dipropionate (EDP, Ovahormone Depot; ASKA Pharmaceutical Co.) at 1.5 mg/body at either 10 or 11 days after the onset of estrus (day 0 = the day of first EDP treatment), and additional EDP (1.5 mg/body) treatments at day 4 and day 7, to establish pseudopregnancy as previously described (Noguchi et al., 2016). After the absence of estrus was confirmed, the recipients were given two intramuscular injections of prostaglandin F2α (PGF2α) as 1.5 mg dionprost (Panacelan Hi; Meiji Seika, Tokyo, Japan), twice within a 24-hour interval at 27 or 30 days after the first administration of EDP.
The recipients were given a single intramuscular injection of eCG (Serotropin; ASUKA Animal Health Co., Ltd., Tokyo, Japan) at 250 IU/body at the same time as the second PGF2α administration. The recipients were also given a single intramuscular injection of hCG (Gonatropin 1000; ASUKA Animal Health Co., Ltd.) at 500 IU/body at 72 hours after the administration of eCG. The transfer of embryos was performed under general anesthesia, through a midline incision at 31 hours (without VPA treatment) or 55 hours (with VPA treatment) after the hCG injection. The same number of SCNT embryos were transferred to the oviducts on each side of the recipient gilts at 4–6 hours (without VPA treatment) or 28–30 hours (with VPA treatment) after activation. Estrus detection was performed 2–3 weeks after embryo transfer. Pregnancy diagnosis was performed ∼4 weeks after embryo transfer, and then fetal survival was determined by using ultrasound.
Microsatellite marker analysis
Genomic DNA was extracted from the piglet, donor cells, and the recipient gilt by using DNeasy Total DNA Kit (Qiagen, Inc., Chatsworth, CA). Genotyping of 12 microsatellite markers, SWR1941, S0038, S0091, SW2108, SW1434, SW2494, SW1378, TNFB, SW1954, SW1681, SWR153, and S0227 on the USDA swine linkage maps (Rohrer et al., 1996) was outsourced (Systembiotics Co., Kanagawa, Japan).
Experimental designs
In experiment 1, miniature pig SCNT embryos derived from intact or transfected CKCs were treated with 0, 4, 8, and 12 mM VPA for 24 hours after activation, and they were examined for in vitro development. In addition, expression of tdTomato and EGFP in blastocysts developed from the transfected CKC-derived SCNT embryos was examined after treatment with VPA.
In experiment 2, microminipig SCNT embryos derived from female kidney cells (adult kidney cells [AKCs] 1) were treated with and without 8 mM VPA for 24 hours after activation, and they were examined for in vitro development.
In experiment 3, microminipig SCNT embryos derived from various donor cells were treated with 8 mM VPA for 24 hours after activation, and they were examined for in vitro development. Cells derived from female and male kidneys (AKCs 1 and 2, respectively); two newborn females (newborn ear cells [NECs] 1 and 2); three newborn males (NECs 3, 4, and 5); and four fetuses (fetal fibroblasts [FFs] 1, 2, 3, and 4) were used as donors.
In experiment 4, microminipig SCNT embryos derived from FFs 1, 2, and 3 were transferred to synchronized gilts after treatment with or without 8 mM VPA for 24 hours after activation, and they were examined for in vivo development.
Statistical analysis
All percentage data were subjected to an arcsin transformation in each replicate. The transformed values and numbers of cells in blastocysts were analyzed by one-way ANOVA, followed by Fisher's protected least significant difference test. A probability of p < 0.05 was considered statistically significant.
Results
Experiment 1
When intact CKCs were used as donors, no significant differences were noted in the cleavage rates (60.4%–73.9%) of embryos among the different concentrations of VPA (Table 1). In contrast, the blastocyst formation rates (14.8%–16.7%) of embryos treated with 4–12 mM VPA were significantly higher (p < 0.05) than those (9.3%) of embryos cultured without VPA treatment. The mean numbers of cells (39.7–46.1 cells) in blastocysts treated with 4–12 mM VPA were not significantly different from those (40.8 cells) in blastocysts cultured without VPA treatment.
CKCs were used as donors. Experiments were repeated five times.
Values with different superscripts are significantly different (p < 0.05).
CKCs, Crawn miniature pig kidney-derived cells; SEM, standard error of the mean; VPA, valproic acid.
Similarly, when transfected CKCs were used as donors, the cleavage rates (37.8%–52.3%) of embryos treated with 4–12 mM VPA were not significantly different from those (47.8%) of embryos cultured without VPA treatment (Table 2). However, the blastocyst formation rates (7.0%–8.9%) of embryos treated with 8–12 mM VPA were significantly higher (p < 0.05) than those (1.0%–2.0%) of embryos treated with 0–4 mM VPA. Among the various concentrations of VPA, no significant differences were noted in the mean numbers of cells (44.0–57.0 cells) in blastocysts.
CKCs transfected with pdOEN-2 and pAQC plasmids were used as donors.
Values with different superscripts are significantly different (p < 0.05).
All blastocysts obtained after treatment with 0–12 mM VPA expressed tdTomato (Fig. 2A, B), indicating that they were derived from SCNT embryos (and not from parthenogenetically activated oocytes) (Table 3). The blastocysts expressing EGFP (Fig. 2C, D) after treatment with 8 mM VPA amounted to 93.3%, and this was significantly higher (p < 0.01) than those after treatment with 0–4 mM or 12 mM VPA (0%–22.2%). Thus, the effects of 8 mM VPA on the in vitro development of microminipig SCNT embryos were examined in the next experiment.
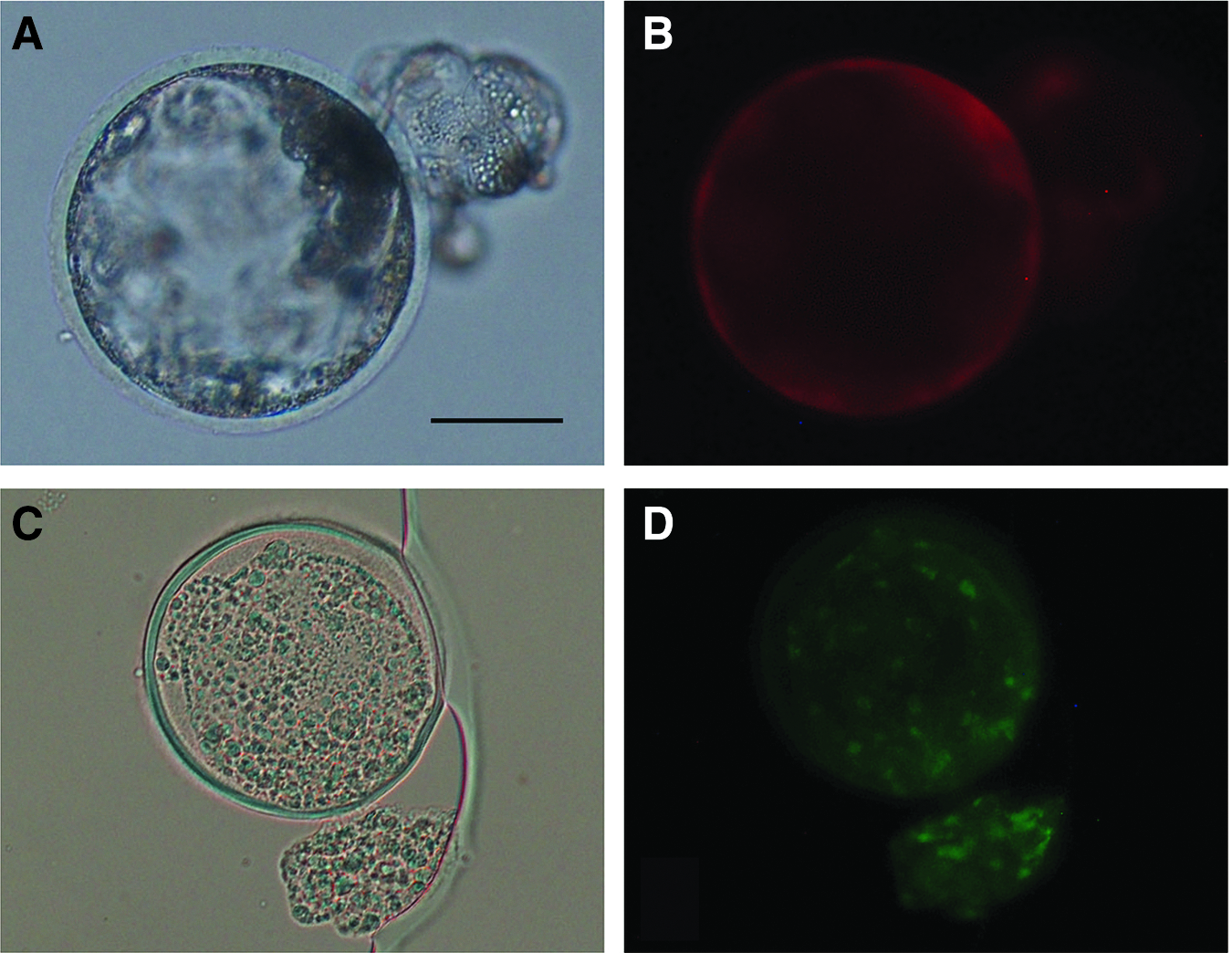
Blastocysts developed from embryos reconstituted with CKCs transfected with pdOEN-2 and pAQC plasmids, and treated with 8 mM VPA for 24 hours, expressing tdTomato-derived
CKCs transfected with pdOEN-2 and pAQC plasmids were used as donors.
Values with different superscripts are significantly different (p < 0.01).
Experiment 2
The presence or absence of VPA treatment did not significantly affect cleavage rates (68.3%–73.8%) of embryos (Table 4). However, the blastocyst formation rate (27.8%) of embryos treated with VPA was significantly higher (p < 0.05) than that (17.1%) of embryos cultured without VPA treatment. The mean numbers of cells (39.5–40.8 cells) in blastocysts were also not affected by VPA treatment. Therefore, SCNT embryos were treated with 8 mM VPA in further experiments.
AKCs were used as donors. Experiments were repeated five times.
Embryos were treated with 8 mM VPA.
Values with different superscripts are significantly different (p < 0.05).
AKCs, adult kidney cells.
Experiment 3
The individual differences in donor cells did not significantly affect the fusion rates of cell-oocyte complexes, the cleavage and blastocyst formation rates of embryos, and the mean numbers of cells in blastocysts in the AKCs and FFs groups (Table 5). In contrast, the fusion rates of cell-oocyte complexes and the mean numbers of cells in blastocysts were significantly affected by the individual differences of donor cells in the NECs group (p < 0.05). Although the fusion rates (67.2%–74.7%) of cell-oocyte complexes, the cleavage rates (73.5%–74.9%) of embryos, and the mean numbers of cells (38.3–42.1 cells) in blastocysts did not differ significantly among the AKCs, NECs, and FFs groups, the blastocyst formation rate (24.2%) of embryos in the FFs group was significantly higher (p < 0.05) than that (16.1%) of embryos in the NECs group. Thus, FFs were used as donors in the next experiment.
Embryos were treated with 8 mM VPA.
Values with different superscripts within the NECs group and each column are significantly different (p < 0.05).
Values with different superscripts within totals are significantly different (p < 0.05).
FFs, fetal fibroblasts; ND, not determined; NECs, newborn ear cells.
Experiment 4
When 162 SCNT embryos derived from FFs 1 (55 embryos), FFs 2 (56 embryos), and FFs 3 (51 embryos) were transferred to a recipient gilt (#1247) without VPA treatment, the gilt did not become pregnant, as determined by ultrasound 29 days after transfer (Table 6). On the other hand, a recipient gilt (#1320) into which 152 SCNT embryos derived from FFs 1 (50 embryos), FFs 2 (55 embryos), and FFs 3 (47 embryos) were transferred after VPA treatment maintained her pregnancy to term and 5 female piglets were delivered via Cesarean section, 113 days after transfer. Three piglets weighing 390, 396, and 398 g were delivered live and showed normal growth (Fig. 3; herein referred to as alive 1, alive 2, and alive 3); however, two piglets weighing 155 and 221 g were stillborn (herein referred to as dead 1 and dead 2). Dead 1 showed multiple anomalies, including hypoplasia of the lumbar vertebrae and hind legs, absence of the sacral vertebrae, anury, anal and pudendal atresia, cranial meningocele, bilateral hydronephrosis, and retroperitoneal hernia. Dead 2 had a cleft palate and exhibited dysraphism of the anterior fontanel.

Cloned microminipigs developed from somatic cell nuclear transfer embryos treated with 8 mM VPA for 24 hours.
FFs 1, FFs 2, and FFs 3 were used as donors.
Embryos were treated with 8 mM VPA.
Genetic analysis confirmed that dead 1, dead 2, alive 1, and alive 2 were identical to FFs 2 at all 12 polymorphic microsatellite loci examined (Table 7). Alive 3 was identical to FFs 1 at all 12 polymorphic microsatellite loci. In addition, dead 1, dead 2, alive 1, and alive 2 lacked all of the alleles detected in the recipient gilt at 5 loci. Alive 3 also lacked the alleles detected in the recipient gilt at 4 loci. These results indicate that the recipient gilt made no genetic contribution to the piglets. On the basis of genetic analysis, it was demonstrated that dead 1, dead 2, alive 1, and alive 2 were derived from FFs 2, and alive 3 was derived from FFs 1.
Discussion
In the present study, we demonstrated that (1) a short duration of treatment with VPA improves reprogramming and/or in vitro development of miniature pig and microminipig SCNT embryos; and (2) microminipig SCNT embryos treated with VPA can be successfully developed to term. As far as we know, this is the first report of the production of cloned microminipigs by SCNT.
The results of the present study indicate that treatment with 8 mM VPA for 24 hours after activation leads to an increase in the rate of blastocyst formation in miniature pig and microminipig SCNT embryos derived from intact and transfected cells. On the other hand, the mean number of cells in blastocysts treated with VPA was not significantly different from that in blastocysts cultured without VPA treatment. These findings suggest that treatment with VPA enhances the ability of SCNT embryos to develop into blastocysts, while maintaining blastocyst quality. These data also support our previous findings on the beneficial effects of VPA treatment on in vitro development of miniature pig SCNT embryos (Miyoshi et al., 2010a). Similar effects of VPA on the in vitro development of pig SCNT embryos have also been reported by other researchers; however, the optimal conditions for VPA treatment as determined by each study are different.
Huang et al. (2011) reported that treatment with 1 mM VPA for 14–16 hours after activation resulted in the highest rate of blastocyst formation in Landrace SCNT embryos. On the other hand, Kang et al. (2013) observed the highest rate of blastocyst formation, when Wuzhishan miniature pig SCNT embryos were treated with 2 mM VPA for 24 hours after activation. Notably, the optimal concentrations of VPA determined in these studies were lower than those determined in the present study. This difference could be attributed to differences in the breeds of donor cells and/or SCNT protocols. Wrenzycki et al. (2001) reported that two different protocols, in which activation is induced at 15 minutes or 3–5 hours after the application of electrical pulses for fusion, alter the expression patterns of developmentally important genes in cattle SCNT embryos. In this context, it is noteworthy that the interval between fusion and activation in the present study was different from those in previous studies.
When we examined the expression of Oct-3/4 promoter-driven EGFP in miniature pig SCNT embryos during development, we observed morulae and early blastocysts mostly at 5 days of culture; however, similar findings were not evident in late blastocysts and hatching blastocysts at 7 days of culture (Miyoshi et al., 2010a). These results suggest that Oct-3/4 activity in miniature pig SCNT embryos is high at the morula to early blastocyst stages, and it is reduced at the late blastocyst to hatching blastocyst stages. Lee et al. (2006) examined the levels of Oct-3/4 transcripts by reverse transcription (RT)-PCR using miniature pig SCNT embryos, and they also reported that although the transcripts were detected at high levels in morulae and blastocysts, their relative amounts were significantly reduced in hatched blastocysts compared with in vivo-developed counterparts. Thus, in the present study, we examined the effects of VPA on expression of the Oct-3/4 promoter gene-driven EGFP in late blastocysts and hatching blastocysts at 7 days of culture. The results of the present study showed that treatment of miniature pig SCNT embryos with 8 mM VPA for 24 hours resulted in prolonged expression of EGFP beyond the late blastocyst stage, indicating that Oct-3/4 activity in these embryos became similar to that in in vivo-developed counterparts.
These results suggest that gene expression in the transfected somatic cell nuclei can be reprogrammed to mimic that of preimplantation embryos, by treating the SCNT embryos with VPA. Improvements in the in vitro developmental ability in the blastocysts of SCNT embryos by VPA could be attributed to prolonged expression of the Oct-3/4 gene, since mouse embryos that lack Oct-3/4 gene expression are unable to form normal blastocysts (Nichols et al., 1998). When miniature pig SCNT embryos were treated with 4 mM VPA for 48 hours in a previous study, only 40.4%–58.5% of those blastocysts expressed EGFP at 7 days of culture (Miyoshi et al., 2010a). Increasing the concentration of VPA to 10 mM or extending the duration of VPA treatment to 72 hours increased the percentage of blastocysts expressing EGFP to 87.5%–90.0%, but it simultaneously reduced the rate of blastocyst formation in SCNT embryos.
In contrast, treatment of SCNT embryos with 8 mM VPA for 24 hours allowed 93.3% of those blastocysts to express EGFP, while maintaining the highest rate of blastocyst formation observed in the present study. Modifying the level of histone acetylation in the transfected somatic cell nuclei at the early stages after activation by a short duration of treatment with a relatively high concentration of VPA might be effective in reprogramming without inhibiting the developmental potential of SCNT embryos. Prolonged exposure to VPA seems to be detrimental to the development after reprogramming of SCNT embryos.
The results of the present study indicate that the type of donor cells affects in vitro development of microminipig SCNT embryos, and among the three types of cells examined, FFs are the most efficient donors. Similar results have been reported in other pig SCNT studies (Lee et al., 2003; Wei et al., 2013). FFs could possibly be highly undifferentiated and, therefore, more amenable to reprogramming than differentiated cells after being transferred into recipient oocytes. In addition, FFs derived from different fetuses seem to have uniform characteristics, compared with NECs, in which both the fusion rate and the mean number of cells in blastocysts differed among different sources. Therefore, the use of FFs as donors might constitute one of the most efficient choices to produce cloned microminipigs by SCNT.
In the present study, we successfully produced cloned microminipigs derived from SCNT embryos treated with VPA. Both Huang et al. (2011) and Kang et al. (2013) reported that VPA treatment enhances in vitro development up to the blastocyst stage of pig SCNT embryos, as described earlier. However, regarding the effects of VPA treatment on in vivo development of SCNT embryos into piglets, their results stood in contrast with each other. In the former study, the developmental rate of SCNT embryos into piglets was significantly improved after treatment with VPA (Huang et al., 2011).
In contrast, the latter reported that VPA treatment does not significantly affect the in vivo development of SCNT embryos; indeed, VPA might actually have a negative effect on cloning efficiency, as 9 out of 11 piglets derived from embryos treated with VPA died within 1–5 days after birth and only 2 piglets survived for more than 3 months. On the other hand, 8 out of 12 piglets derived from untreated embryos survived beyond 3 months (Kang et al., 2013). This disparity could be attributed to differences in the genetic background of the donor cells, as Huang et al. (2011) and Kang et al. (2013) used donor somatic cells derived from common porkers and inbred miniature pigs, respectively. In mice, the effects of VPA treatment on the development of SCNT embryos into cloned offspring seem to be strain specific. VPA improved the development of SCNT embryos that had been reconstituted with cells derived from B6CBAF1 (C57BL/6J × CBA/J) mice (Costa-Borges et al., 2010), whereas it did not affect cloning efficiency in the BD129F1 (B6D2F × 129/Sv) strain (Ono et al., 2010).
When microminipig SCNT embryos were transferred without VPA treatment, no piglets were obtained in the present study. We believe that gene expression in most microminipig SCNT embryos differs from that in preimplantation embryos, but it can be reprogrammed to mimic that of preimplantation embryos by treating with VPA in the same way as miniature pig SCNT embryos. However, we cannot determine whether VPA improves cloning efficiency in microminipigs at present, because only a small number of recipient gilts were used in the present study. Nevertheless, no piglets died immediately after birth, an observation that is not consistent with those reported by Kang et al. (2013). This suggests that VPA might not have an adverse effect on the viability of cloned microminipigs. Microsatellite marker analysis indicated that four out of five cloned piglets obtained in the present study were derived from the same donor cell line (FFs 2). It has been reported that different cell lines derived from the same source show a considerable degree of variation in cloning efficiency (Kurome et al., 2013). Thus, the use of FFs 2 as donor cells for SCNT would facilitate the efficient production of cloned microminipigs.
In the present study, two out of five cloned piglets developed from SCNT embryos treated with VPA were stillborn and showed several types of phenotypic abnormalities. It is unclear whether this phenomenon can be attributed to treatment with VPA, because stillborn and/or deformed piglets are generally obtained at a relatively high frequency with SCNT (Schmidt et al., 2015). Further investigation will be required to clarify this point.
In conclusion, we established an efficient SCNT protocol for the production of cloned microminipigs. This protocol would be effective in producing GM microminipigs that can be used as a model for human diseases, based on their shared similarities in anatomical and physiological characteristics with humans. In addition, these animals can be kept in relatively small spaces, without the need for much labor.
Footnotes
Acknowledgments
The authors thank Drs. Kazuyuki Ohbo (School of Medicine, Yokohama City University, Yokohama, Japan) and Roger Tsien (Department of Pharmacology, University of California, La Jolla, CA) for providing the pGOF-18 plasmid and tdTomato cDNA, respectively. They wish to express their gratitude to the staff of the Kagoshima City Meat Inspection Office and Meat Center Kagoshima (Kagoshima, Japan) for supplying pig ovaries. They thank Yozo Nagao for his technical support. The present study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (Nos. 25450475 and 16K08085 to K.M., and 24580411 to M.S.).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.