
Abstract
Abstract
Constant levels of homeobox transcription factor Prox1 expression are required throughout the life of lymphatic endothelial cells (LECs) to maintain their differentiated identity. Recent studies have demonstrated that using human LECs for cell transplantation therapy may improve secondary lymphedema in a nude rat model. However, the application is currently limited by the low yield of LECs. In this study, Prox1 was overexpressed in human adipose tissue-derived stem cells (hADSCs) by using the transfection of lentiviral vectors to induce the differentiation of hADSCs to LECs. After 14 days of Prox1 overexpression, flow cytometry analysis found that the expression of LEC-specific markers such as Podoplanin and VEGFR3, along with the endothelial cell (EC) marker CD31, on Prox1-overexpressed hADSCs was significantly increased; however, the expression of mesenchymal stem cell markers, such as CD29, CD44, and CD90, was substantially reduced. In addition, the mRNA levels of the LEC-specific markers, such as Prox1, Podoplanin, LYVE1, and VEGFR3, in Prox1-overexpressed hADSCs were significantly increased at day 7 and maintained a continuously increased expression level for 28 observation days, according to real-time reverse transcriptase–polymerase chain reaction results. Western blotting and immunofluorescence staining results further confirmed that overexpression of Prox1 in hADSCs significantly increased the protein levels of Podoplanin, LYVE1, and VEGFR3, as well as those of the EC markers such as VWF and CD144, at day 14. Moreover, these differentiated cells were found to form tube-like structures in matrigel, measured by the tube formation assay. These findings suggested that overexpression of Prox1 in hADSCs successfully induced the differentiation of hADSCs into stable lymphatic endothelial-like cells. This study achieved a long-lasting expression of Prox1 in lymphatic endothelial-like cells, and it provided a potentially useful approach for developing novel therapies for limb lymphedema and lymphatic system-related diseases.
Introduction
L
It has been shown that mesenchymal stem cells (MSCs) isolated from the bone marrow incubated with lymph-inductive media and purified VEGF-C resulted in the expression of LEC markers in vitro (Conrad et al., 2009). However, human bone marrow-derived MSCs (BMSCs) are acquired from a painful and invasive surgical procedure. Compared with BMSCs, adipose tissues are more widely available and easily obtained by less traumatic methods such as liposuction (Gimble, 2003; Gimble and Guilak, 2003a, 2003b). Researchers have confirmed that human adipose tissue-derived stem cells (hADSCs) are capable of differentiating into lymphatic endothelial-like cells (Hwang et al., 2011; Yan et al., 2011; Yang et al., 2015); the current methods mainly utilize VEGF-C to induce this differentiation.
For example, Hwang et al. (2011) reported that hADSCs injected with a VEGF-C-impregnated hydrogel sheet in a mouse hindlimb lymphedema model resulted in the regeneration of LECs at the transplanted site. Yan et al. (2011) reported that VEGF-C-pretreated hADSCs led to a markedly increased expression of Podoplanin and Prox1 in hADSCs. Yang et al. (2015) used VEGF-C156 s to promote hADSC differentiation and obtained a certain number of lymphatic endothelial-like cells. However, these methods were not able to establish continuous stimulation, because the inductive effect of VEGF-C was temporary (Yan et al., 2011).
Previous reports showed that changes in the genetic expression level were sufficient to initiate a reprogramming cascade, leading to the dedifferentiation of LECs (Bixel and Adams, 2008; Johnson et al., 2008). Therefore, whether this kind of differentiation effect induced by VEGF-C can be sustained has not been confirmed. Furthermore, exogenous VEGF-C treatment may have undesired and yet-to-be-determined side effects, such as inducing the formation of hyperplastic but dysfunctional lymphatics (Goldman et al., 2005; Rutkowski et al., 2006). For these reasons, developing a method to continuously and safely acquire and maintain cells with an LEC phenotype is crucial for the cell transplantation therapy for lymphedema.
The homeodomain protein Prox1 plays an essential role in specifying the LEC phenotype during embryogenesis (Wigle et al., 2002). The first lymphatic sprouts that normally bud out from the anterior cardinal vein were arrested in Prox1-deficient embryos and failed to develop further (Hong et al., 2002; Wigle and Oliver, 1999; Wigle et al., 2002). In addition, a constant level of Prox1 activity in the embryonic and mature lymphatic vasculature was crucial to maintain their phenotypic identity (Johnson et al., 2008). A previous study showed that an LEC phenotype could be acquired by way of Prox1 overexpression. For example, Petrova et al. (2002) found that overexpression of Prox1 in venous endothelial cells (ECs) was sufficient to activate the transcription of lymphatic-specific genes (VEGFR3 and Podoplanin). However, venous ECs are also a limited resource and are difficult to acquire; thus, this method would hardly be useful for the clinical treatment of lymphedema.
To date, the inducing effects of Prox1 on the differentiation of hADSCs have not been reported. Therefore, in this study, we investigated whether overexpressing Prox1 in hADSCs by means of lentiviral transfection could potentially induce the differentiation of hADSCs into LECs. LEC-specific phenotypes were subsequently evaluated by examining the cell proliferation, gene/protein expression of LEC (or EC) markers, and the functional assessment of ECs.
Materials and Methods
Collection, isolation, and culture of hADSCs
Protocols for the handling of human tissue and cells were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine. Human adipose tissues were donated by the patients for research purposes only with written informed consent. Human adipose tissues were harvested from the abdomen or inner thigh of patients by standard liposuction procedures. The samples were washed extensively with chloramphenicol once and phosphate-buffered saline (PBS) twice and then digested by 0.075% collagenase type I (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 h with vigorous shaking. Equal volumes of Dulbecco's modified Eagle medium (DMEM; HyClone, Logan City, UT) containing 10% fetal bovine serum (FBS, BioSum, South America) were added to neutralize the collagenase activity.
The cell suspension was centrifuged at 1200g for 10 min, and the pellet was resuspended in DMEM containing 10% FBS (BioSum, South America) and antibiotics (penicillin 100 U/mL, streptomycin 0.1 mg/mL) and incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. These primary cells were defined as passage 0 and were passaged once 80%–90% confluency was reached. The cells were cultured to passage 3 before use in all the experiments in this study.
Identification of hADSCs
The cells were verified by flow cytometry and examined for multiple lineage differentiation as requested by the International Society for Cellular Therapy. For flow cytometry analysis, hADSCs of passage 3 were incubated with monoclonal PE-conjugated antibodies for CD29, CD31, CD44, CD45, CD90, Podoplanin, and VEGFR3. The detailed steps are described in flow cytometry analysis.
For adipogenesis assessment, the cells were incubated in adipogenic differentiation medium, which consisted of DMEM culture medium that was supplemented with 0.5 mM 1-methyl-3-isobutylxanthine, 100 mM indomethacin, 1 mM dexamethasone, and 10 μL/L insulin (all from Sigma-Aldrich). This medium was exchanged every 3 days. After 21 days of differentiation, the accumulation of neutral lipid droplets in the cytoplasm was confirmed by positive oil red O staining that was observed under an inverted phase-contrast microscope (Olympus, Japan).
For osteogenic differentiation, the cells were incubated in DMEM culture medium containing 10−8 M dexamethasone, 0.3 g/L
Lentiviral vector construction and transfection
All lentiviral vectors used for this research were purchased by Shanghai SunBio Medical Biotechnology Co., Ltd. According to the manufacturer's protocol, to acquire the lentiviral vectors used for this experiment, the recombinant miRNA expression vectors and Lentivirus Package plasmid mix (Supplementary Fig. S1) were co-transfected in human embryonic kidney 293T cells with Lipofectamine 2000 (Invitrogen). hADSCs were inoculated on 24-well culture dishes at a density of 1 × 104 cells/cm2. Lentiviral vectors encoding the Prox1 and GFP genes, or GFP gene only, were added once the cells reached 60% confluence in the 24-well culture dish; these were defined as the Prox1/GFP group or GFP group, respectively. Multiplicity of infection (MOI) was determined to be 30 for achieving the optimal transfection activity. The titer of the lentiviral vector was 2.28 × 108 TU/mL, and the amount per well was calculated as follows: number of cells × MOI/titer of lentiviral vector. Polybrene (Sigma) was added for enhanced transfection activity (1:1000).
All of the lentiviruses used in this study were generated with puromycin resistance, and all of the transfected cells were selected by puromycin treatment (1.5 μg/mL). In addition, before the start of this experiment, cell cycle synchronization was verified by flow cytometry on an FACScan flow cytometer (Becton-Dickinson, San Jose, CA). Moreover, to guarantee the validity of the experiment, before each assay in this study, including the cell proliferation assay, flow cytometry analysis, real-time reverse transcriptase–polymerase chain reaction (RT-PCR), western blot, immunofluorescence staining, and tube-forming assay, was carried out, GFP+ cells were selected by using FACS technique. The transfection efficiency was calculated as the number of fluorescence-marked cells/total cell number × 100%. The hADSCs cultured with DMEM containing FBS and antibiotics only were defined as the blank control group (DMEM + FBS group).
Flow cytometry analysis
Flow cytometry was used to identify the following groups: cells before transfection, cells 14 and 28 days after transfection in the Prox1/GFP group, and cells 14 days after transfection in the GFP group. The cells were dissociated by using Accutase (Sigma) and passed through a cell strainer (40-μm pore size) to obtain single cells. The immunophenotype was defined by using monoclonal antibody reagents that were directed against CD29, CD31, CD44, CD45, CD90, Podoplanin, and VEGFR3 (all from the BioLegend company with PE conjugation) in staining media (4% FBS in PBS) at room temperature for 30 minutes. For CD29, CD31, CD44, CD90, and Podoplanin, 10–15 μL antibody was added per one million cells that were incubated in 100 μL staining media. For CD45 and VEGFR3, 5 μL antibody was added per one million cells in 100 μL staining media. The cells were then washed twice with PBS, fixed with 4% formaldehyde, and examined on an FACScan flow cytometer (Becton-Dickinson, San Jose, CA).
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8 kit, Dojindo, Kumamoto, Japan) was used to evaluate cell proliferation between the Prox1/GFP group and DMEM + FBS group. After 7 days of transfection, the cells were inoculated on 96-well plates in 100 μL DMEM containing 10% FBS and antibiotics (penicillin 100 U/mL, streptomycin 0.1 mg/mL) at a density of 1 × 104 cells/mL per well and then cultured for 1–8 days before analysis. One hundred microliter DMEM + FBS + antibiotics only was added to the two groups on cell adherence. At the end of each desired checkpoint, 10 μl CCK-8 kit solution was added to each well and then incubated at 37°C for 2 h. Thereafter, the media were collected and subjected to absorbance measurement on a microplate reader (Molecular Devices, Sunnyvale, CA) at 490 nm wavelength. The assay was performed in five parallel wells.
Immunofluorescence staining
Dissociated cells from the Prox1/GFP group and GFP group were cultured on glass coverslips for 2 days at post-transfection day 14 and then fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.1% Triton X-100 (Fisher Scientific) in PBS solution for 5 minutes, blocked with 1% goat serum for 15 minutes, and incubated with primary antibody at 4°C for 12 h. The primary antibodies included rabbit anti-human LYVE1 (1:200; AngioBio), rabbit anti-human VWF (1:200; AngioBio), mouse anti-human Podoplanin (1:100; AngioBio), and rabbit anti-human CD144 (1:200; Abcam).
Thereafter, the cells were incubated with secondary antibody at 37°C for 1 h. Goat anti-rabbit IgG (1:150; AngioBio), goat anti-rabbit IgG (1:200; Abcam), and goat anti-mouse IgG (1:100; AngioBio) were used as secondary antibodies (all were conjugated to Alexa Fluor® 594 [red]). The cell nuclei were stained with DAPI solution (1:1000; Invitrogen) for 2 minutes at room temperature. Between each step, the cells were rinsed extensively three times for 5 minutes each. The results were examined and photographed with a fluorescence microscope (Olympus, Japan).
Real-time reverse transcriptase–polymerase chain reaction
For the RT-PCR detection of lymphatic-specific markers, total RNA was extracted by homogenization with Trizol Reagent (Invitrogen, Carlsbad, CA) at post-transfection days 7, 14, 21, and 28. Reverse transcription was performed by using 2 μg of total RNA as a template and the reagents supplied in the One-Step RT-PCR kit (TaKaRa, Japan) according to the manufacturer's instructions. RT-PCR analysis of the Prox1, Podoplanin, LYVE1, VEGFR3, and β-actin mRNA was performed by using the Mx3000P RT-PCR system (Stratagene). The RT-PCR conditions were as follows: denaturation at 95°C, 15 s; primer annealing at 60°C, 12 s; and elongation at 72°C, 12 s for 40 cycles. The oligonucleotide primer sets selected from human genes are shown in Table 1.
Western blot analysis
At post-transfection day 14, cells from the Prox1/GFP group and DMEM + FBS group were lysed in RIPA buffer containing protease inhibitors (Promega Corporation, Madison, WI) for 30 minutes on ice, cleared by centrifugation, and the supernatants were collected for further analysis. The protein extracts were denatured by heat at 100°C for 5 min and electrophoretically separated on a 12% SDS-PAGE (Bio-Rad). The proteins were transferred to PVDF membranes, blocked with 5% milk/Tris buffered saline, incubated with rabbit anti-human Podoplanin antibody (1:400; AngioBio), rabbit anti-human LYVE1 antibody (1:400; AngioBio), rabbit anti-human VEGFR3 antibody (1:500; AngioBio), or mouse anti-human β-tubulin (1:400; AngioBio) in TBST buffer at 4°C overnight, and then incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse IgG (1:10,000; AngioBio) secondary antibodies at room temperature for 2 hours. The image was scanned with a GS 800 Densitometer Scanner (Bio-Rad). A mouse anti-human β-tubulin polyclonal antibody (1:10,000; AngioBio) was used as an internal control.
Tube formation assay
At post-transfection day 14, suspended cells (4 × 104/well) from the Prox1/GFP group and GFP group were added to 24-well plates that were pre-coated with Matrigel™ (BD Biosciences) at post-transfection day 14. Serum-free medium was added to the wells. The formation of tube-like structures was observed at 12 hours after the addition of cells under a fluorescence microscope (Olympus, Japan).
Data analysis
The data are presented as the mean ± standard error of the mean. Each experiment was repeated at least three times. To determine the significance of the differences between study groups, a one-way ANOVA statistical test was performed. A p value <0.05 was considered statistically significant.
Results
Identification of ADSCs
ADSCs exhibited a typical fibroblast-like morphology and expanded easily when cultured in DMEM culture medium in vitro (Fig. 1A). The cultured cells could also be successfully trans-differentiated into adipocytes and osteocytes, confirmed by using oil red O staining and alkaline phosphatase staining (Fig. 1B–D). The result of the flow cytometry analysis showed that before transfection (shown as pre-transfection in Fig. 2A), the typical MSC markers CD29 (98.8% ± 0.46%), CD44 (95.8% ± 1.1%), and CD90 (98.7% ± 0.35%) were strongly expressed in the majority of cells, whereas the EC marker CD31 (0.5% ± 0.01%), white blood cell marker CD45 (0.1% ± 0.01%), and LEC markers Podoplanin (0.7% ± 0.02%) and VEGFR3 (0.4% ± 0.01%) were expressed at low levels in the majority of cells (Fig. 2A). These data corroborated previous studies (Gimble and Guilak, 2003a, 2003b) and demonstrated that our experimental method successfully produced a highly pure source of hADSCs.
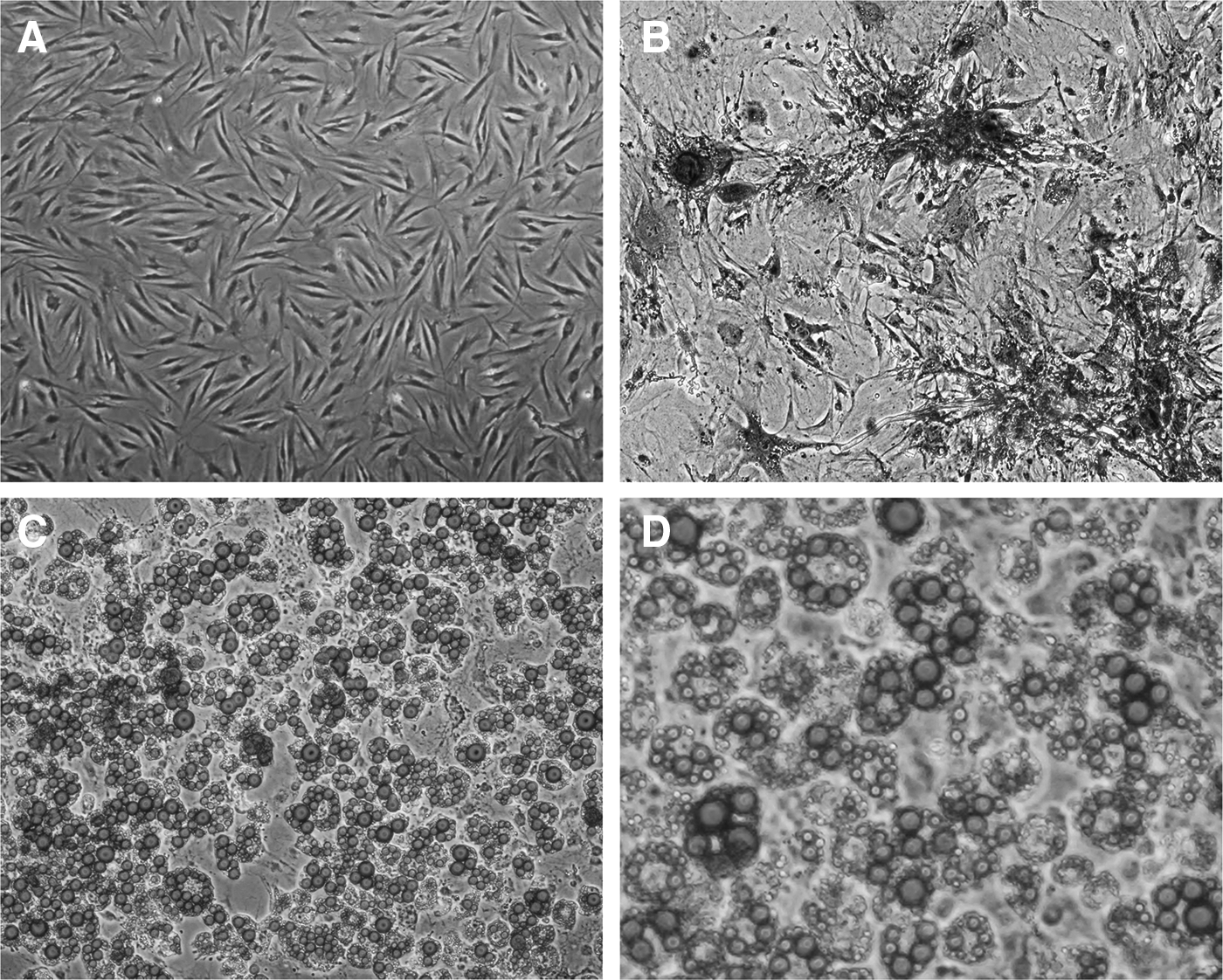
Cultured hADSCs were able to differentiate toward adipogenic and osteogenic lineages in vitro.
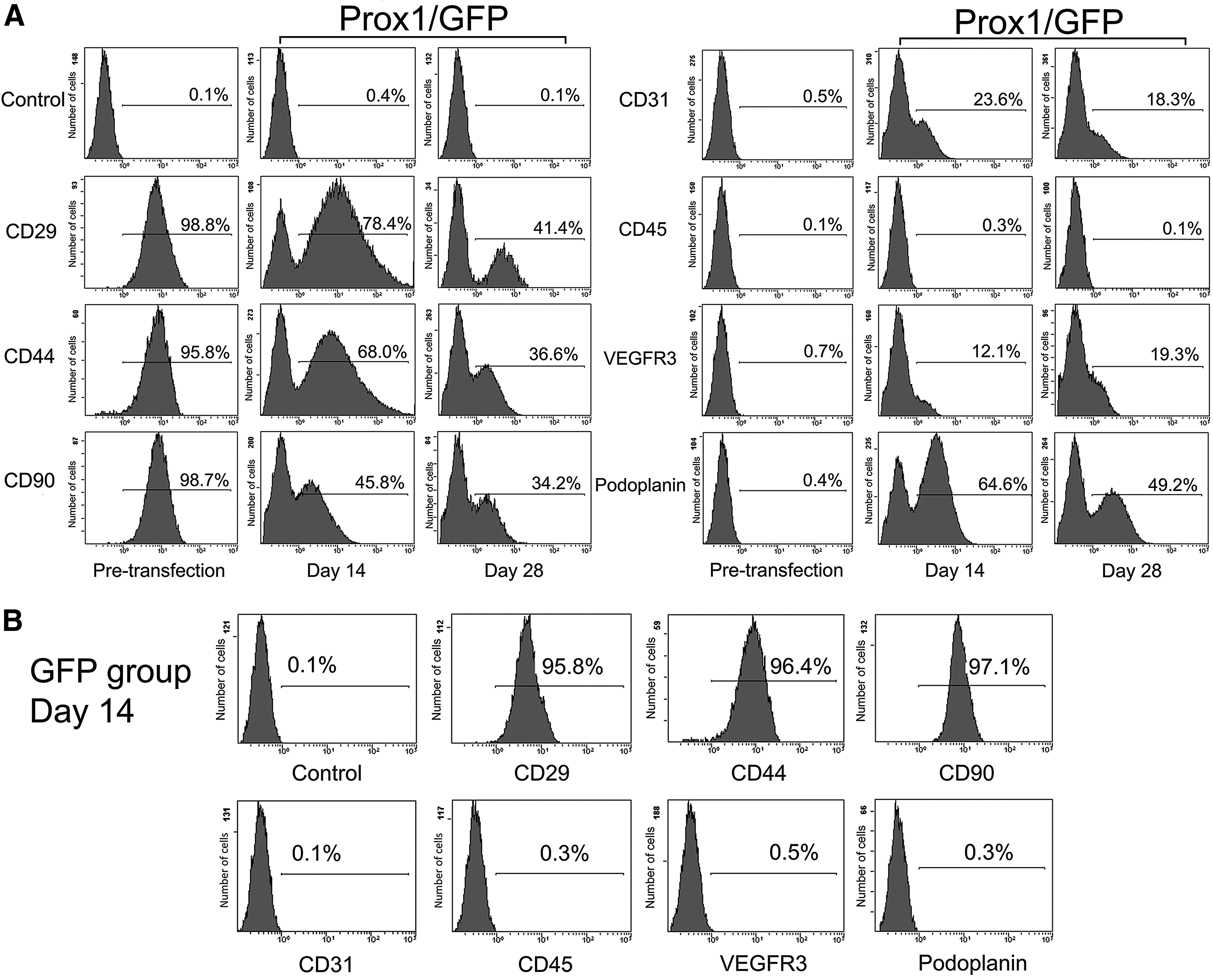
Flow cytometry analysis showed the changes in the expression of cell-surface antigens on hADSCs both before and after transfection.
Flow cytometry detected an increased expression of LEC-specific markers but a decreased expression of MSC-specific makers
Flow cytometry was used to evaluate the expression of cell-surface antigens on the hADSCs both before and after Prox1 overexpression. According to the aforementioned results, before lentiviral vector transfection (pre-transfection), the cultured hADSCs were confirmed to positively express CD29, CD44, and CD90, and confirmed to express a low level of CD31, CD45, Podoplanin, and VEGFR3 in the majority of cells (Fig. 2A). Surprisingly, after 14 days of Prox1 overexpression in hADSCs, flow cytometry revealed a decreased expression of CD29 (78.4% ± 3.4%, p < 0.01), CD44 (68.0% ± 2.9%, p < 0.01), and CD90 (45.8% ± 4.1%, p < 0.01), and a significantly increased expression of Podoplanin (64.6% ± 4.6%, p < 0.01), VEGFR3 (12.1% ± 1.1%, p < 0.01), and CD31 (23.6% ± 2.9%, p < 0.01) in the Prox1/GFP group, compared with the result before transfection (Fig. 2A).
Furthermore, a continuously decreased expression of CD29 (41.4% ± 2.9%, p < 0.01), CD44 (36.6% ± 4.1%, p < 0.01), and CD90 (34.2% ± 4.6%, p < 0.01) and still a high-level expression of Podoplanin (49.2% ± 5.2%, p < 0.01), VEGFR3 (19.3% ± 3.5%, p < 0.01), and CD31 (18.3% ± 3.5%, p < 0.01) were detected at day 28 in the Prox1/GFP group, compared with the result before transfection (Fig. 2A). However, the results in the GFP group at day 14 had no difference compared with the results before transfection (p > 0.05). The expression of CD29, CD44, and CD90 was still high, whereas the expression of CD31, VEGFR3, and Podoplanin was low in the majority of hADSCs (Fig. 2B).
Overexpression of Prox1 promoted the morphologic transition of hADSCs but did not affect their proliferation
The morphologic transition of hADSCs in the Prox1/GFP group from a fibroblast-like appearance to a typical EC-specific cobblestone-like appearance could be clearly observed at day 14, whereas hADSCs in the GFP group remained in a fibroblast-like appearance (Fig. 3A). Transfection efficiency was evaluated at post-transfection days 2, 7, and 14. The results in the GFP group were 49.2% ± 5.2% (day 2), 70.1% ± 4.3% (day 7), and 84.3% ± 3.4% (day 14); whereas in the Prox1/GFP group, the results were 47.1% ± 4.0% (day 2), 68.7% ± 3.9% (day 7), and 82.7% ± 3.6% (day 14), with no significant difference between the GFP group and Prox1/GFP group at each time point (p > 0.05) (Fig. 3B). Statistical analysis of the CCK-8 assay results showed that p > 0.05 when comparing the Prox1/GFP group and DMEM + FBS group at each time point, suggesting that overexpression of Prox1 did not affect the proliferation of hADSCs (Fig. 3C).
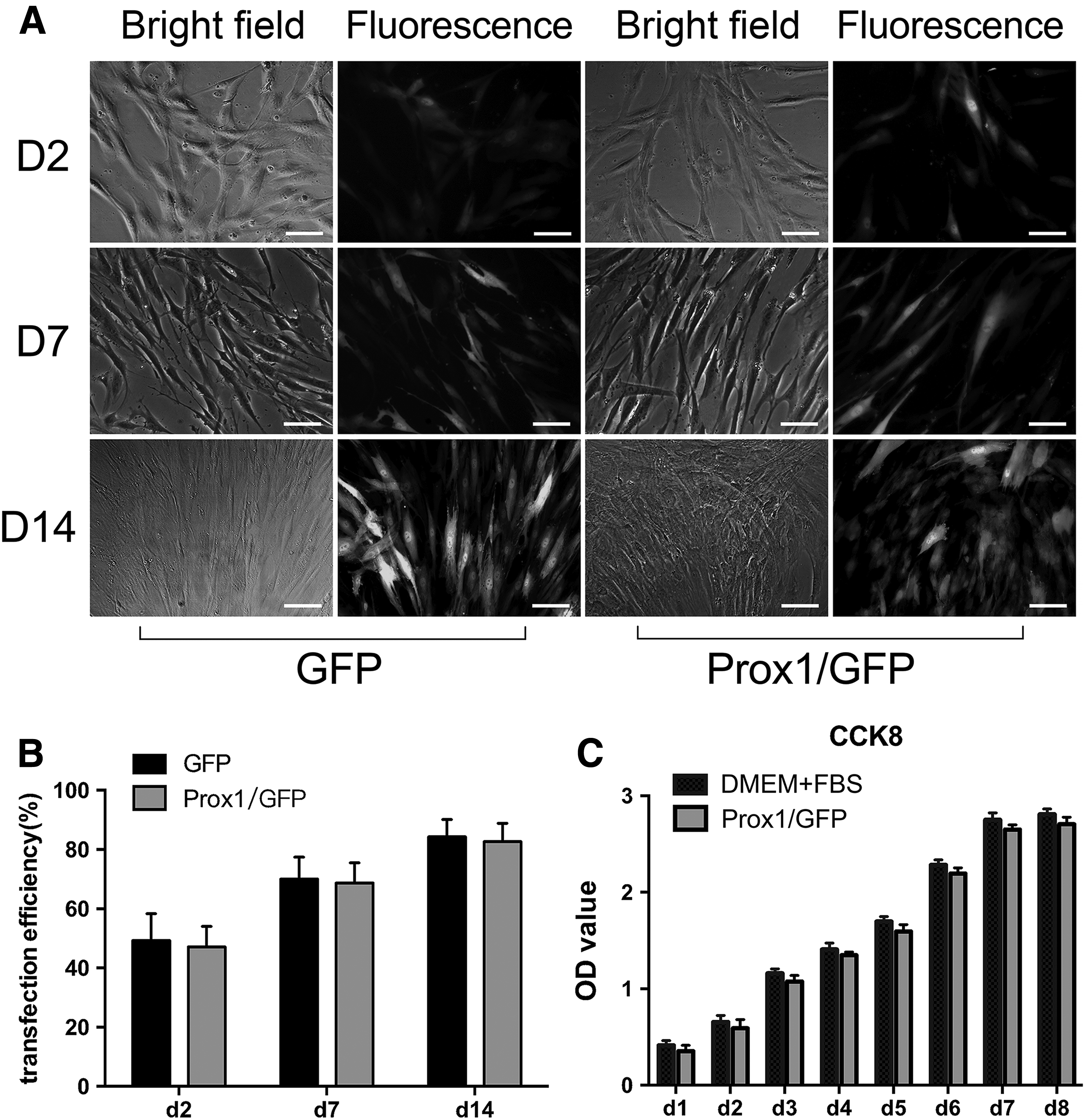
Cell morphological observation and proliferation assay.
Overexpression of Prox1 promoted hADSCs to acquire a lymphatic endothelial-specific phenotype
RT-PCR was performed to evaluate the gene expression profile of Prox1-overexpressed hADSCs at post-transfection days 7, 14, 21, and 28. The results showed that, compared with the DMEM + FBS group, the mRNA levels of Prox1, Podoplanin, LYVE1, and VEGFR3 in the Prox1/GFP group increased by 1.25 ± 0.06-fold (p < 0.05), 1.21 ± 0.03-fold (p < 0.05), 1.41 ± 0.03-fold (p < 0.05), and 1.35 ± 0.04-fold (p < 0.01) at day 7, respectively; 7.2 ± 0.28-fold (p < 0.01), 1.66 ± 0.17-fold (p < 0.05), 1.9 ± 0.12-fold (p < 0.01), and 1.99 ± 0.14-fold (p < 0.01) at day 14, respectively; 4.53 ± 0.28-fold (p < 0.01), 2.26 ± 0.16-fold (p < 0.01), 2.43 ± 0.13-fold (p < 0.01), and 3.1 ± 0.17-fold (p < 0.01) at day 21, respectively; and 2.51 ± 0.16-fold (p < 0.01), 2.43 ± 0.16-fold (p < 0.01), 2.64 ± 0.21-fold (p < 0.05), and 2.79 ± 0.25-fold (p < 0.05) at day 28, respectively (Fig. 4A–D).
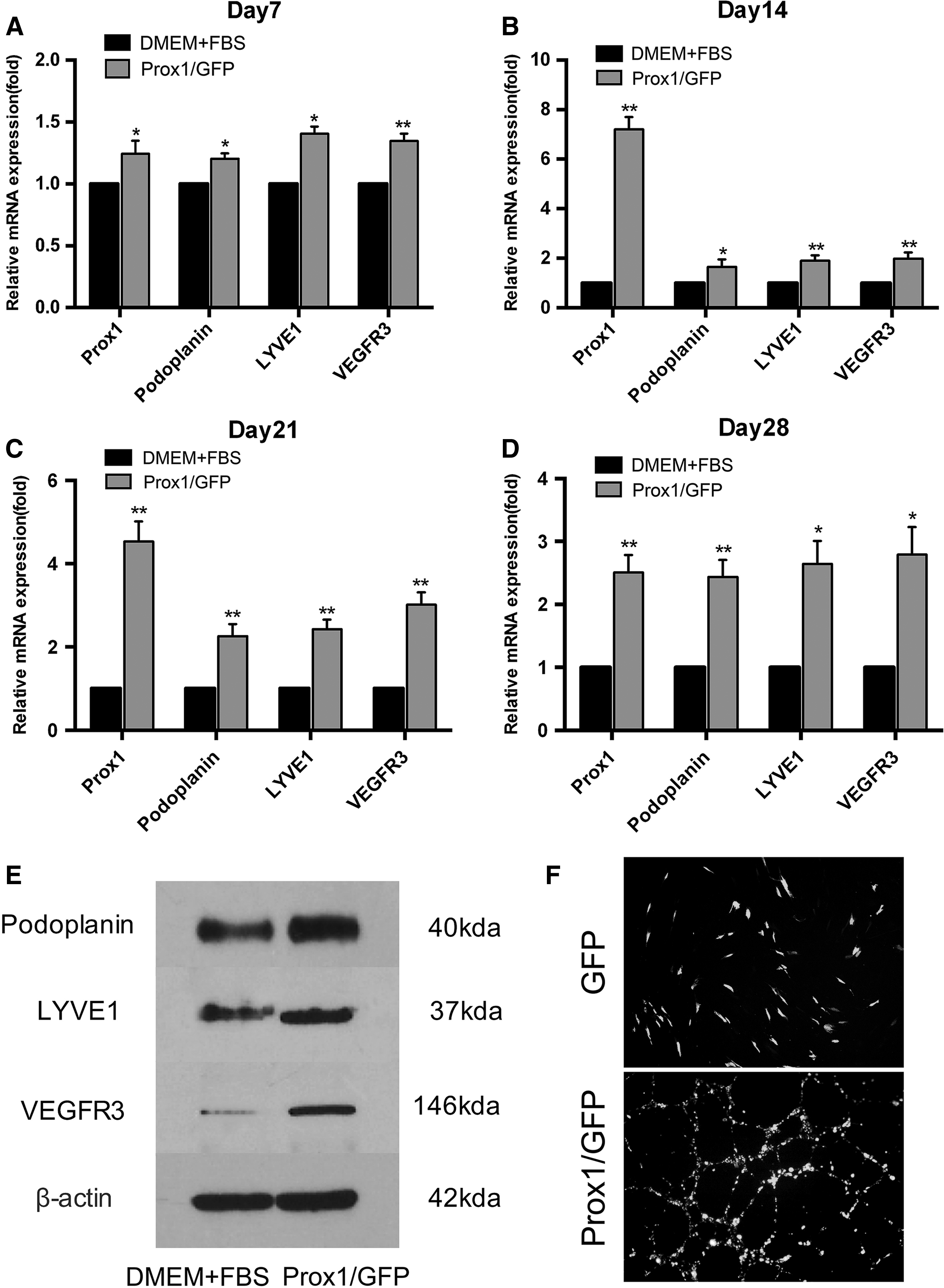
Overexpression of Prox-1 promoted hADSCs to acquire a lymphatic endothelial-specific phenotype.
In addition, western blot analysis revealed a markedly increased protein expression of Podoplanin, LYVE1, and VEGFR3 at day 14 in the Prox1/GFP group, compared with the DMEM + FBS group (Fig. 4E). Furthermore, after 14 days of Prox1 overexpression, the tube formation assay showed that Prox1-overexpressed cells incubated in matrigel for 12 hours were found to form tube-like structures, whereas cells from the GFP group remained irregular, suggesting that these differentiated cells acquired EC-specific characteristics (Fig. 4F). Moreover, the protein expression levels of Podoplanin and LYVE1, and the EC-specific markers VWF and CD144, were also evaluated by means of immunofluorescence staining.
In the Prox1/GFP group, the protein expression of Podoplanin, LYVE-1, VWF, and CD144 was strongly positive at day 14 (Fig. 5). In addition, as CD144 was strongly expressed in the differentiated cells, we also found obvious junctions formed between the cells, as shown in Figure 5B, which further identified these differentiated cells as lymphatic endothelial-like cells. However, the protein expression of Podoplanin, LYVE1 and VWF, and CD144 in the GFP group remained low.
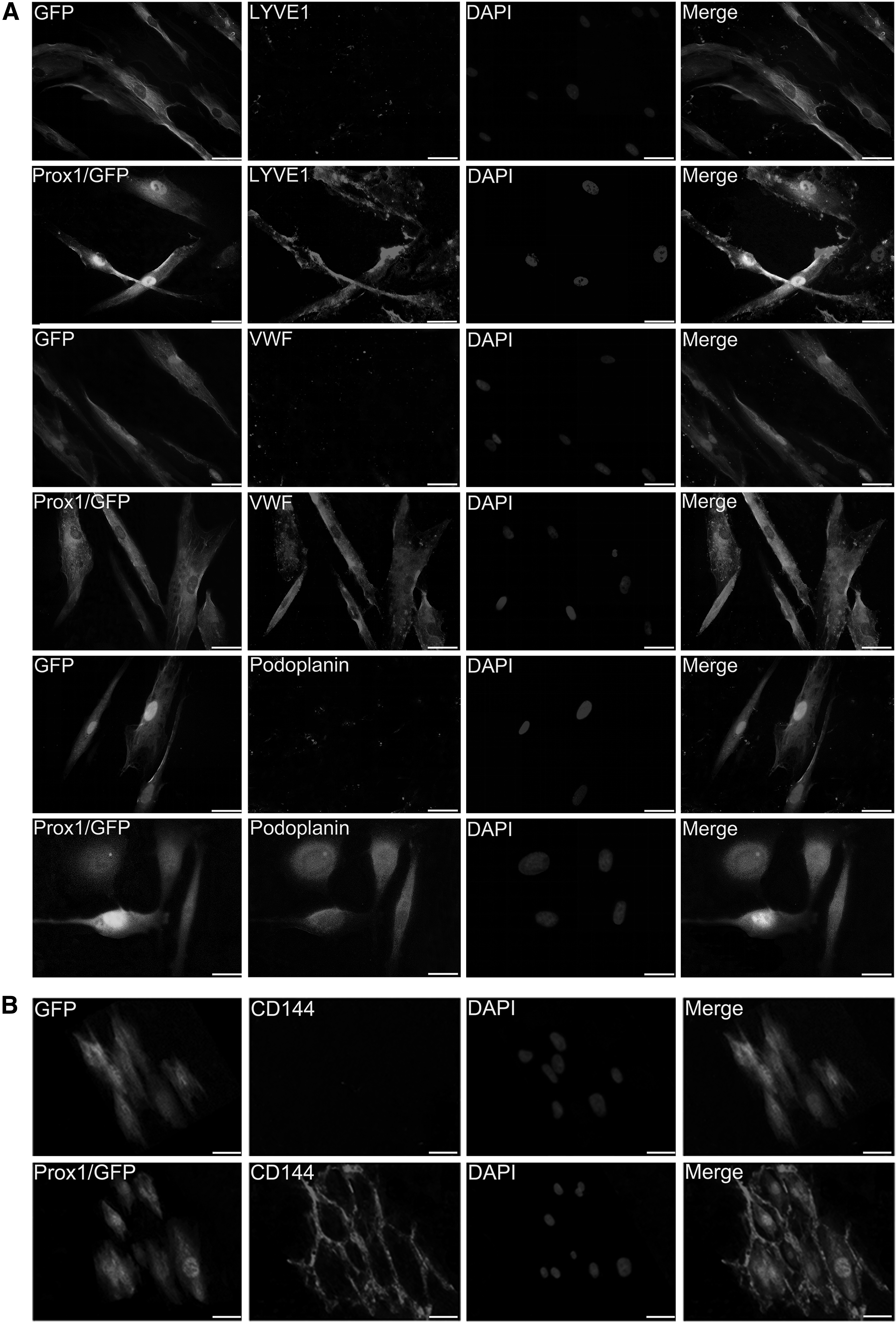
Immunofluorescent staining of transfected hADSCs.
Discussion
It is generally believed that ADSCs originate from the mesoderm, with the potential to differentiate toward adipocytes, chondrocytes, osteoblasts, and myocytes (Baer and Geiger, 2012). Recent studies confirmed that ADSCs have the potential to differentiate into ECs (Mizuno, 2009; Zuk et al., 2001). However, few studies have focused on the differentiation of ADSCs toward lymphatic endothelial-like cells. In this study, we showed that overexpression of Prox1 promoted the expression of LEC-specific makers, such as Podoplanin, LYVE1, and VEGFR3, in differentiated hADSCs. These differentiated cells coincided with the characteristics of ECs with the ability to form tube-like structures in matrigel, as well as the ability to express the intercellular junction marker, CD144.
At the same time, we found that overexpression of Prox1 suppressed the hADSC-specific phenotype but it did not affect cell proliferation. These results confirmed that Prox1 was the key regulator through the whole life of LECs. In addition, it was demonstrated for the first time that the differentiation of hADSCs into LECs could be achieved via a gene overexpression-induced manner. Thus, hADSCs might serve as suitable seed cells for the tissue engineering of lymphatic vessels in vitro and potentially in the later reconstruction of lymphatic ducts after lymphedema in vivo.
Furthermore, we showed that an LEC phenotype induced by Prox1 overexpression was more stable than that induced by stimulation with VEGF-C. As previously reported, VEGF-C is an important factor contributing to the differentiation of ADSCs toward lymphatic endothelial-like cells (Hwang et al., 2011; Yan et al., 2011; Yang et al., 2015), but its treatment was not able to sustain the LEC phenotype for a long time. Lee et al. (2010) found that stimulation of BMSCs with VEGF-C in vitro resulted in a temporary expression of Prox1 for a few days, which was completely lost at day 7. Thus, it was reasonable to assume that differentiated lymphatic endothelial-like cells required continuous stimulation to maintain a lymphatic phenotype. As a comparison, our study found that the expression of Prox1 in differentiated cells was significantly increased since day 7 and was maintained at a high expression level for 28 observation days. To explain this difference, it was thought, presumably, that the exogenous Prox1 gene that integrated into the chromosome of hADSCs via transfection of the lentiviral vector helped to achieve a long-lasting expression of Prox1 (Anastasov et al., 2016).
Interestingly, flow cytometry showed that overexpression of Prox1 in hADSCs for 14 days led to a significant upregulation of the expression of typical LEC markers, such as VEGFR3 and Podoplanin, but a significant downregulation of the expression of ADSC-specific markers, including CD29, CD44, and CD90, suggesting that these Prox1-overexpressed cells began to differentiate toward LECs in response to Prox1. To explain this phenomenon, we speculated that constant expression of Prox1 in hADSCs played an important role in promoting the LEC phenotype by modulating multiple signaling cascades, which helped to activate LEC-specific gene transcription but repressed hADSC-specific gene transcription (Johnson et al., 2008; Oliver and Srinivasan, 2008).
For example, we found that overexpression of Prox1 in hADSCs resulted in the increased expression of VEGFR3, a receptor tyrosine kinase that is specific for the lymphatic endothelium and essential for proper lymphatic growth and function (Karkkainen and Petrova, 2000). Since a previous study confirmed that ADSCs might stably and continuously secrete multiple lymphangiogenic cytokines, including VEGF-C (Yan et al., 2011), we speculated in this study that the upregulated expression of VEGFR3 activated by Prox1 and its interaction with VEGF-C secreted by ADSCs may be an important signaling pathway that is involved in the establishment of LEC identity (Coso et al., 2012).
In addition, we found that overexpression of Prox1 in hADSCs led to an increased expression of Podoplanin, a transmembrane mucin type O-glycoprotein specifically expressed on LECs (Breiteneder-Geleff et al., 1997, 1999). This finding was supported by studies from Hong et al. and Johnson et al., who found that ectopic expression of Prox1 in primary human LECs upregulated the expression of Podoplanin (Hong et al., 2002), whereas knockdown of Prox1 caused the loss of Podoplanin (Johnson et al., 2008); it was suggested that Prox1 could functionally bind to the 5′ regulatory sequence of Podoplanin and trigger downstream gene expression (Pan et al., 2014). However, using VEGF-C as a stimulator for ADSCs or MSCs also resulted in upregulation of Podoplanin expression (Conrad et al., 2009; Yan et al., 2011). Thus, Prox1 may not be the only regulator of Podoplanin, and another regulatory mechanism may involve VEGF-C, which needs further investigation.
Another finding in our study was an increased expression of the lymphatic-specific marker LYVE1 (Banerji et al., 1999), which was detected for 28 observation days. However, previous studies reported that stimulation of ADSCs or BMSCs with VEGF-C in vitro resulted in no expression or only temporary expression of LYVE1 (Lee et al., 2010; Yan et al., 2011). It is possible that Prox1 may regulate the expression of LYVE1, as reported by Wigle et al. (2002), that Prox1-null murine ECs, normally destined to become LECs, failed to properly express LYVE1. Consequently, the result confirmed that Prox1 was an essential factor for the expression of LYVE1, and constant expression of Prox1 in hADSCs was crucial for the establishment of LEC identity.
In conclusion, this study showed that Prox1 played a vital role in the formation of the LEC phenotype by promoting the expression of LEC-specific markers. More importantly, it was the first time that a Prox1-overexpression approach was used to induce the differentiation of hADSCs into stable lymphatic endothelial-like cells. Importantly, this method could achieve a long-lasting expression of Prox1 and avoided the potential side effects caused by exogenous VEGF-C treatment, which may have profound significance in the development of a novel cell therapy for limb lymphedema and lymphatic system-related diseases. However, further studies on Prox1-overexpressed hADSCs, especially regarding the effectiveness and safety in therapeutic models, are necessary to evaluate.
Footnotes
Acknowledgments
This work was supported by the Innovation Program of Shanghai Municipal Education Commission (13ZZ090), the National Natural Science Foundation of China (81371700, 81501571), and the Natural Science Foundation of Shanghai (16ZR1419700).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.