
Abstract
Abstract
For successful cartilage tissue engineering, the ability to generate a high number of chondrocytes in vitro while avoiding terminal differentiation or de-differentiation is critical. The ability to accomplish this by using the abundant and easily sampled costal cartilage could provide a practical alternative to the use of articular cartilage and mesenchymal stem cells. Chondrocytes isolated from pig costal cartilage were expanded in either serum-free medium with FGF2 (SFM) or fetal bovine serum-containing medium (SCM), under either high (21%) or low (5%) oxygen conditions. Overall, chondrocytes cultured in SFM and low oxygen (Low-SFM) demonstrated the highest cell growth rate (p < 0.05). The effect of passage number on the differentiation status of the chondrocytes was analyzed by alkaline phosphatase (AP) staining and real-time PCR for known chondrocyte quality markers. AP staining indicated that chondrocytes grown in SCM had a higher proportion of terminally differentiated (hypertrophic) chondrocytes (p < 0.05). At the mRNA level, expression ratios of ACAN/VCAN and COL2/COL1 were significantly higher (p < 0.05) in cells expanded in Low-SFM, indicating reduced de-differentiation. In vitro re-differentiation capacity was assessed after a 6-week induction, and chondrocytes grown in Low-SFM showed similar expression ratios of COL2/COL1 and ACAN/VCAN to native cartilage. Proteomic analysis of in vitro produced cartilage indicated that the Low-SFM condition most closely matched the proteomic profile of native costal and articular cartilage. In conclusion, Low-SFM culture conditions resulted in improved cell growth rates, reduced levels of de-differentiation during expansion, greater ability to re-differentiate into cartilage on induction, and an improved proteomic profile that resembles that of in vivo cartilage.
Introduction
A
Injured cartilage possesses limited regeneration ability and produces a fibrous cartilage scar that is composed of a different type of extracellular matrix (ECM). Hence, clinically, tissue replacement therapy by using autologous chondrocyte implantation (ACI) harvested from a patient's own cartilage might be a more suitable approach. However, there is a limit to the availability and size of the biopsy that can be obtained from articular cartilage. As a result, chondrocytes need to be expanded in vitro by sequential passaging, and this expansion has been shown to induce de-differentiation and phenotypic changes, leading to chondrocytes that no longer perform as desired (Darling and Athanasiou, 2005; Goessler et al., 2004; Thorogood et al., 1986). Further, chondrocytes harvested from older or diseased joint cartilage have a low proliferation capacity, thus making the expansion of chondrocytes difficult (Dehne et al., 2009; Wenger et al., 2006).
These limitations of in vitro chondrocyte expansion can be overcome by the modification and optimization of culture conditions such as utilization of serum-free medium containing growth factors (Giannoni et al., 2005; Mandl et al., 2004b; Martin et al., 1999; Pei et al., 2002) and the use of reduced oxygen levels (Adesida et al., 2012a, 2012b; Foldager et al., 2011; Robins et al., 2005). Particularly, the addition of fibroblast growth factor 2 (FGF2) to conventional chondrocyte culture medium containing fetal bovine serum (FBS) enhanced both cell proliferation and cartilage formation during re-differentiation (Jakob et al., 2001; Martin et al., 1999).
However, the use of serum (FBS) is problematic due to batch-to-batch variation, resulting in difficulties in obtaining consistent results and complicating Food and Drug Administration (FDA) approval for clinical applications (Wright et al., 1988). Therefore, standardized and optimized culture systems, which can yield high-quality chondrocytes, are strongly needed to assess the suitability of nonarticular tissue-derived chondrocytes for ACI (Sayed et al., 2010). Previously, costal cartilage showed its potential as an alternative chondrocyte source for ACI (Isogai et al., 2006; Murphy et al., 2013).
Further, animal studies have shown that costal cartilage can provide higher cell yield and expansion of chondrocytes when compared with articular cartilage (Lee et al., 2007; Szeparowicz et al., 2006). Also, the expanded costal chondrocytes were successfully transplanted and engrafted into articular lesions, independent of the age of the animals used (Szeparowicz et al., 2006), thus reassuring costal cartilage as an alternative source of chondrocytes for the patients for ACI.
In this study, our goal is to establish complete serum-free culture chondrocytes that would allow robust in vitro expansion of chondrocytes while retaining their ability to generate high-quality cartilage on induction, and to determine whether costal chondrocytes, an easily sampled and abundant source of chondrocytes, could be used as an alternative to MSCs and articular chondrocytes for cartilage tissue engineering.
To test these possibilities, we utilized swine costal cartilages and their chondrocytes as a model system to be able to translate the results in vivo by using a highly suitable and amenable large animal model of cartilage repair that closely resembles humans. Using porcine costal chondrocytes, we compared the effects of a hypoxic environment of 5% oxygen with a normoxic environment of 21% oxygen, and a conventional culture system containing FBS with a defined serum-free medium containing fibroblast growth factor (FGF2).
To optimize the serum-free culture conditions, we reasoned that the serum-free culture media widely used in mouse and human embryonic stem cell culture, which rely on knockout serum replacement (KSR) as a substitute for FBS, might be beneficial to the growth of chondrocytes (Amit et al., 2000) (Cheng et al., 2004). The hypoxic environment was used to simulate the avascular in vivo conditions of cartilage tissue, which is estimated to be between 2% and 7% oxygen (Brighton and Heppenstall, 1971). Defined serum-free media containing FGF2 were used to both abolish the use of FBS, facilitating regulatory approval, and take advantage of the ability of basic fibroblast growth factor (FGF2) to reduce de-differentiation and maintain chondrocytic potency.
The effects of low oxygen, FGF2, and their synergic effect on cell proliferation and de-differentiation were monitored during in vitro expansion, and their re-differentiation capacity was analyzed by pellet culture and by proteomic analysis of the resulting cartilage.
Materials and Methods
Chondrocyte isolation
Chondrocytes were isolated from costal cartilage that was obtained from three 2–5 month-old pigs. To isolate primary chondrocytes, the cartilage was thoroughly minced with scalpel blades into fine fragments (less than 1 mm2). Minced cartilage fragments were washed in Dulbecco's phosphate buffered saline (DPBS; Cellgro, Manassas, VA) and digested with 1.2% type II collagenase (Worthington, Lakewood, NJ) at 37°C for 6 hours. The cell suspensions were then filtered through cell strainers (70 μm; BD Falcon, Franklin Lakes, NJ) to obtain single cells. Cells were washed three times in DPBS and re-suspended in culture medium containing either FBS or FGF2. The number of total cells and viable cells were measured by Trypan blue staining (Sigma, St. Louis, MO) and by counting with a Bright-Line™ Hemacytometer (Sigma; average cell yield: 470,000 cells per gram).
Chondrocyte culture
Viable cells were plated into 6- and 24-well plastic culture dishes at a density of 15,000 cells/cm2. Cells were cultured at 37°C under 5% CO2 in combinations with either 21% O2 (High) or 5% O2 (Low), and either FBS-containing medium (SCM) or serum-free medium containing FGF2 (SFM). For hypoxia culture, Heracell™ 150i Tri-gas incubator (Thermo Scientific) was utilized to control oxygen level. This resulted in four different combinations (High-SCM, High-SFM, Low-SCM, and Low-SFM). SFM was composed of Dulbecco's modified Eagle medium (DMEM; Cellgro) supplemented with 10% KSR (Life Technologies, Breda, Netherlands), 100 ng/mL FGF2 (re-combinant human; Stemgent, Cambridge, MA), and 50 ug/mL gentamycin (Cellgro). SCM medium consisted of DMEM with 10% FBS and 50 ug/mL gentamycin.
Chondrocytes expanded in SFM were incubated in 50:50 mixtures of SCM and SFM to promote cell adhesion in the culture dishes for the first 12 hours after seeding. After the cells had adhered, they were washed three times with DPBS to get rid of residual FBS and fed with fresh SFM. Medium was replaced every other day. Cell doubling time was calculated by the formula: T × ln2/ln (Nf/Ni), where the T is the incubation time, Nf is the cell number counted at the end of the incubation time, and Ni is the initially plated cell number. Chondrocytes were plated at 1500/cm2 at all passages; then, cell numbers were counted at the end of each passage (2–3 days) to calculate cell doubling time.
In addition, we compared cellular expansion yield and induction of hypertrophic chondrocytes between seven different batches of FBS to confirm that the results were not due to poor-quality FBS, and to ensure that the results were not lot dependent. The SFM always demonstrated highly enhanced cell yield and the lowest level of hypertrophic chondrocytes when compared with all seven batches of SCM (data not shown). Seven different batches of FBS were purchased from four different vendors: Cellgro (Lot. 08CG09 and FB11554), Sigma (Lot. 8G005), Serum source (Charlotte, NC; Lot. FBU10519), and Gemini (Sacramento, CA; Lot. A74C05Z, A93C00C, and A89C05C).
All of the results shown were generated by using the batch of FBS that demonstrated the highest cell yield and lowest levels of hypertrophic chondrocytes in comparison with other batches of FBS (Cellgro, Lot. FB11554).
Alkaline phosphatase staining
Chondrocytes were treated with VECTASTAIN ABC-AP kit (VECTOR Laboratories, Burlingame, CA) according to the manufacturer's protocol. After alkaline phosphatase (AP) staining, pictures of each treatment were taken (20 × magnification; AZ100, Nikon, Japan) and three microscopic fields were randomly selected. The AP-stained surface area was measured and quantitated by using ImageJ (version 1.46; NIH, Bethesda, MD) from the selected fields. Each measurement score was normalized to the number of cell counts of the corresponding treatment.
Pellet culture for in vitro re-differentiation
Passage 3 chondrocytes from each treatment were used for pellet culture to study their chondrogenic potential as previously described (Estes et al., 2010). Briefly, expanded chondrocytes were trypsinized and re-suspended in re-differentiation induction medium. The induction medium was composed of DMEM supplemented with a 1/100 dilution of insulin, transferrin, and selenious acid (ITS+; Becton Dickinson, insulin, transferrin, selenious acid, bovine serum albumin, and linoleic acid), 10 ng/mL insulin-like growth factor 1 (MBLI, Woburn, MA), Dexamethasone (100 nM, Sigma), 10 ng/mL transforming growth factor beta 3 (TGF-β3, MBLI), and 10 ng/mL bone morphogenetic protein 6 (BMP-6, MBLI).
As an induction control, incomplete induction medium containing DMEM with 10% FBS, ITS+ (1:100) and Dexamethasone (100 nM) without TGF-β3 and BMP-6, was used. Overall, 250,000 cells were used to make a pellet in a 15-mL tube and the pellet was incubated for 6 weeks. Media were changed every other day with gentle agitation to prevent the pellet from adhering to the tube wall. After 6 weeks of induction, pellets were harvested and used for RNA extraction, histology, and mass spectrometry (MS).
Histological analysis
Chondrocyte pellets (n = 3 for each treatment) and native costal cartilage samples (from three different animals) were washed with DPBS and fixed in 4% paraformaldehyde followed by paraffin embedding. Three sections from each pellet were stained with hematoxylin and eosin and Alcian blue to identify GAG accumulation. To quantify the proportion of the GAG production per microscopic field, the intensity of Alcian blue was measured by collecting images using a Nikon AZ100 microscope and NIS-Elements software (Nikon) followed by analysis with ImageJ software. Alcian blue staining was separated from the background by using the “Color Deconvolution” plugin (Ruifrok and Johnston, 2001), with a threshold setting (threshold = 5) to have all Alcian blue positive pixels selected for analysis. The area of selected pixels is measured in three different zone layers (Zone 1, Zone 2, and Zone 3) to evaluate the homogeneity of produced GAGs in the pellets.
Real-time polymerase chain reaction (PCR) analysis
Total RNA was isolated from three independent chondrocyte cultures (at P1, P2, and P3), chondrocyte pellets (n = 3 for each treatment), and native costal cartilage (from three separate animals) with EZ-10 Total RNA miniprep kit (Bio Basic, Ontario, Canada) following the manufacturer's protocol. Isolated RNA was treated with TURBO DNase (New England Biolabs, Ipswich, MA) according to the manufacturer's instructions. cDNA was synthesized with the AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies, Santa Clara, CA) by using an oligo (dT) primer. Relative gene expression was quantified by iTaq™ SYBR Green Kit (Biorad, Hercules, CA) on an ABI step one plus system (Life Technology).
Conditions for real-time RT-PCR were as follows: 95°C, 15 mins; 40 cycles (Ct ranged from 15 to 30) (95°C, 15 s; 60°C, 5 s; 72°C, 30 s); and 72°C 2 mins, followed by melting curve analysis (90 cycles, start at 50°C with 0.5°C increments). Each reaction was performed in triplicate. The relative expression was calculated by normalization with GAPDH expression using the Pfaffl method (Pfaffl, 2001). Primer sets for real-time polymerase chain reaction (PCR) and their amplification efficiencies are listed in Table 1.
Correlation coefficiency.
PCR efficiency.
PCR, polymerase chain reaction.
Statistical analysis
Real-time PCR results were analyzed by using one-way ANOVA followed by Bonferroni and Tukey's honest significant difference (HSD) post-test. SAS 9.2 software was used for statistical analysis of the data. All data were expressed as mean ± standard error of mean, and significance was set at p < 0.05.
Sample preparation for MS
Costal, articular, and ear cartilage was obtained from three pigs from 1 to 3 months of age. Samples were minced followed by pulverization in liquid nitrogen. Fifty milligram of powdered cartilage from the three donors was pooled to make 150 mg total of each tissue type and was, subsequently, used for protein extraction. Protein samples were prepared as previously described (Onnerfjord et al., 2012). Briefly, proteins were extracted by adding 3 mL of chaotropic extraction buffer (4 M GdnHCl, 50 mM NaAc, 100 mM 6-aminocaproic acid, 5 mM benzamidine, 5 mM N-ethylmaleimide, pH 5.8) followed by 24 hours of incubation on a rotating shaker at 4°C.
Protein extracts were collected by centrifugation at 13,200 g for 30 min at 4°C followed by three washes with chaotropic extraction buffer. Samples were run on a 4%–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (Biorad) gel followed by gel fixation (50% MeOH, 10% Acetate) and coomassie blue staining with GelCode Blue Stain Reagent (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. The entire lane of each sample was cut into five slices, and each slice was minced into 1 × 1 mm pieces and transferred into 1.5 mL tubes. Gel pieces were de-stained with 50:50 100 mM NH4HCO3: acetonitrile solution for 1 hour. Protein in-gel digestion was performed as previously described (Shevchenko et al., 2006) followed by protein purification with C18 ZipTip (Millipore, Billerica, MA) according to the manufacturer's protocol.
MS analysis
All the procedures of database preparation and MS were performed at the North Carolina State University MS facility. Nano-liquid chromatography-MS was performed with LTQ Orbitrap XL (Thermo Scientific), and the raw sequence data were utilized to identify proteins from the primary sequence database by using the search algorithm MASCOT (Matrix Science, Boston, MA) at a 1% false discovery rate. MASCOT results were further analyzed with ProteoIQ software (NuSep, Bogart, GA) for relative quantification. Label-free quantitative proteomic approaches were applied by normalization with normalized spectral abundance factor (Zybailov et al., 2007) at a significant threshold of p < 0.05 compared with the reference sample (articular cartilage). A hierarchical clustering map of protein expression was generated with JMP Genomics 5 (SAS, Cary, NC) by using log2 relative expression ratio of the entire proteome data obtained from ProteoIQ analysis.
Results
Porcine chondrocyte isolation and expansion
We examined the effects of oxygen level (High vs. Low) and serum-free medium with FGF2 compared with serum-containing medium (SCM vs. SFM) on in vitro expansion of chondrocytes. Chondrocytes grown in SCM exhibited a chondrocyte-like polygonal or rounded shape, whereas cells in SFM had a more fibroblast-like shape (Fig. 1). Oxygen level did not have an influence on morphological changes during passages. The cells expanded in SCM at both oxygen levels became larger during extended passages, suggesting hypertrophic differentiation of chondrocytes, whereas the cells in SFM did not exhibit morphological changes until P7.
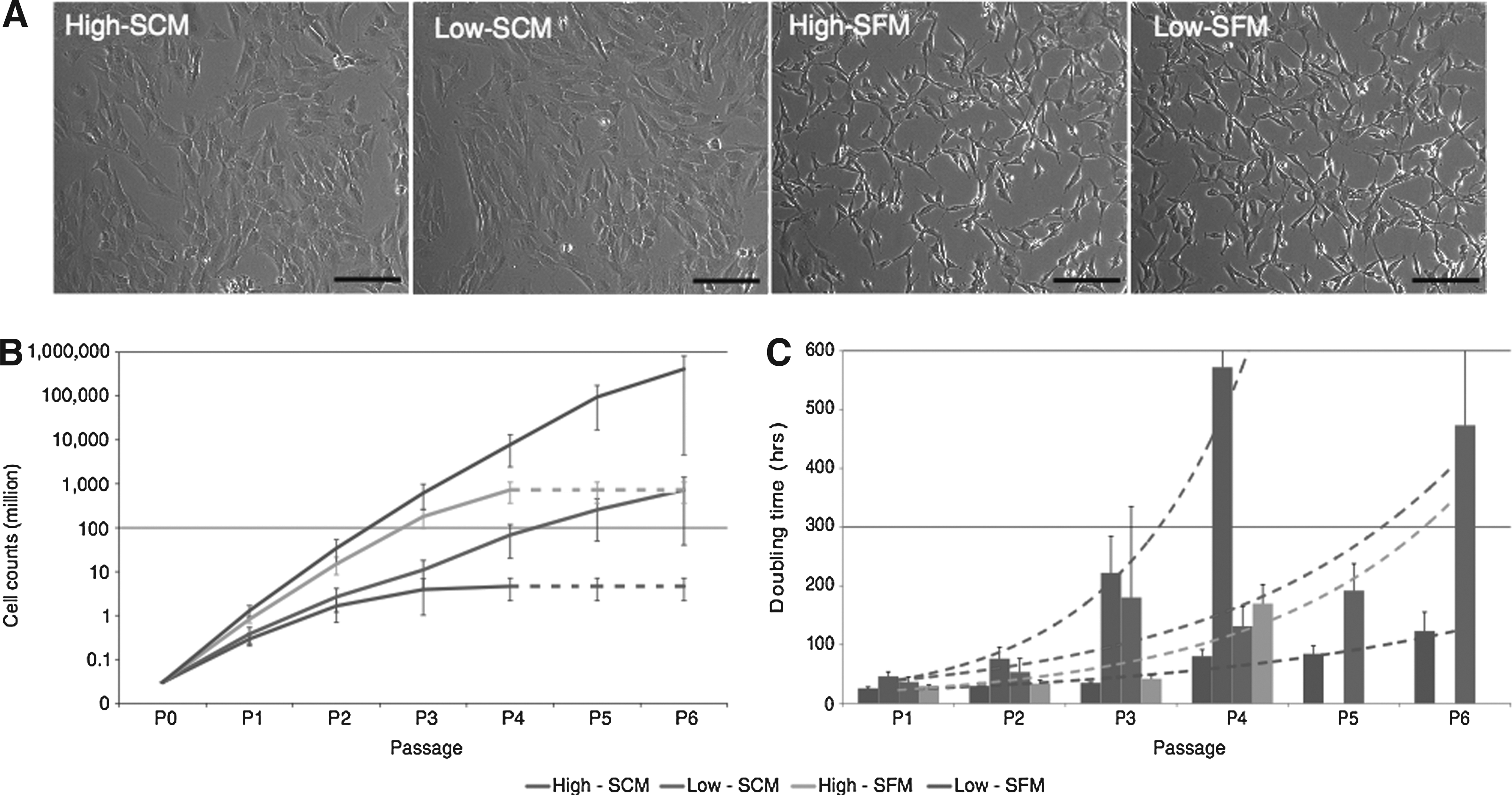
Morphological changes and growth rate of chondrocytes during in vitro expansion.
Both low oxygen level and SFM culture increased total cell counts (Fig. 1), low oxygen promoted maintenance of proliferation rates, and expansion in high oxygen resulted in a decreased growth rate in earlier passages as shown by a rapid increase in doubling time at P3 (High-SCM) or P4 (High-SFM) (p < 0.05, Fig. 1). Cells grown in SFM had higher cell counts compared with SCM groups (p < 0.05, High-SFM vs High-SCM and Low-SFM vs. Low-SCM). Cells cultured in high oxygen stopped proliferating at passage 4 in both serum-containing and serum-free media (Fig. 1).
Hypertrophic differentiation during in vitro expansion
In vitro expanded cells were stained with AP to visualize the proportion of hypertrophic chondrocytes at each passage (P1, P2, and P3), and the fold increase in area occupied by AP-stained cells was calculated. Cells cultured in SFM had less AP-positive hypertrophic chondrocytes in both high and low oxygen levels (Fig. 2). In addition, the combination of low oxygen and SFM resulted in minimal hypertrophic chondrocytes during expansion (Fig. 2). Hypertrophic chondrocytes increased in each passage in all conditions, especially at passage 3; however, Low-SFM always demonstrated the lowest level (p < 0.05) of AP-positive cells in all passages and treatments (Fig. 2).
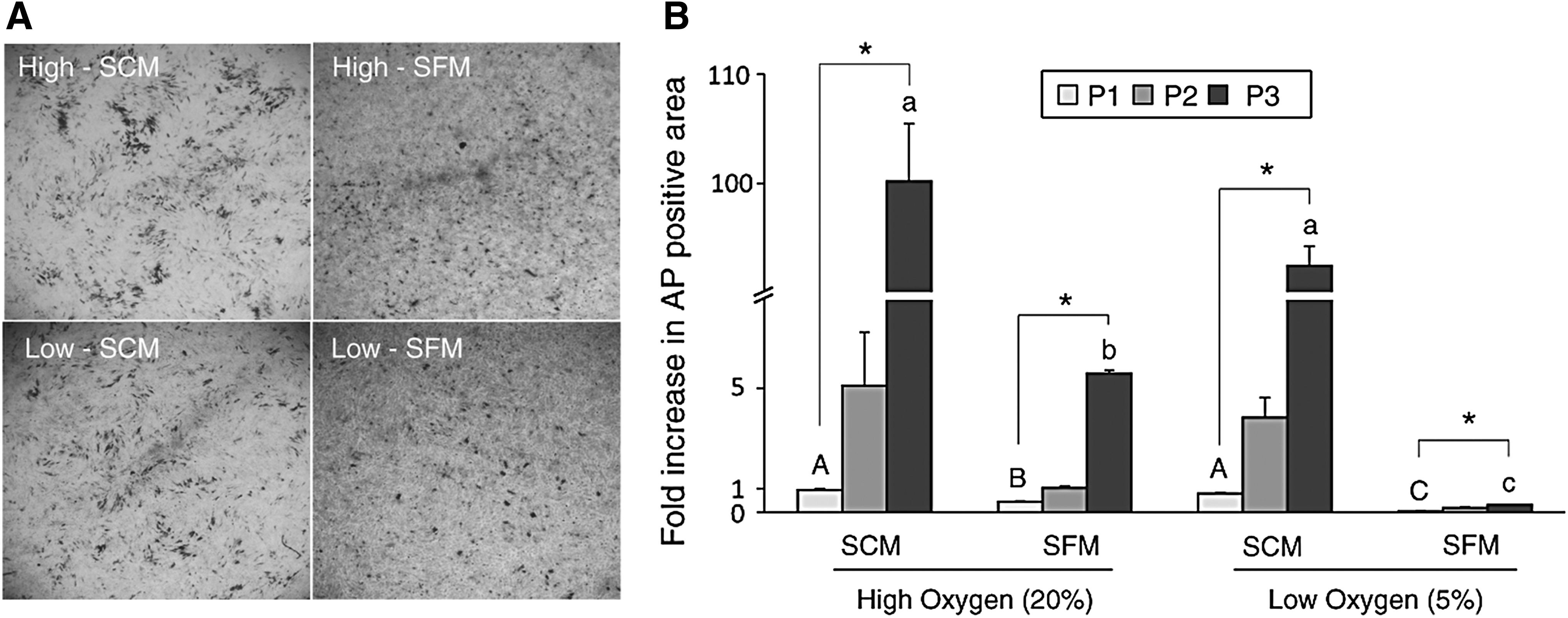
Alkaline phosphatase (AP) staining analysis of in vitro expanded chondrocytes.
Marker expression of in vitro expanded chondrocytes
Chondrocytes cultured in Low-SFM had the highest ratio of ACAN/VCAN and COL2/COL1 expression (p < 0.05); however, the ratios of both ACAN/VCAN and COL2/COL1 significantly declined after the first passage in all four treatments (p < 0.05, Fig. 3). This decrease was due to a decrease in ACAN expression and an increase in VCAN (Fig. 3). At the same time, COL2 expression decreased and COL1 expression increased (Low-SFM) or was maintained during passages P1 through P3 (High-SFM) (Fig. 3). The transcription factor RUNX2 showed a high level of expression in SFM at both oxygen levels (p < 0.05, High-SFM and Low-SFM) (Fig. 3), but expression decreased after passaging and did not show a significant difference at P3.
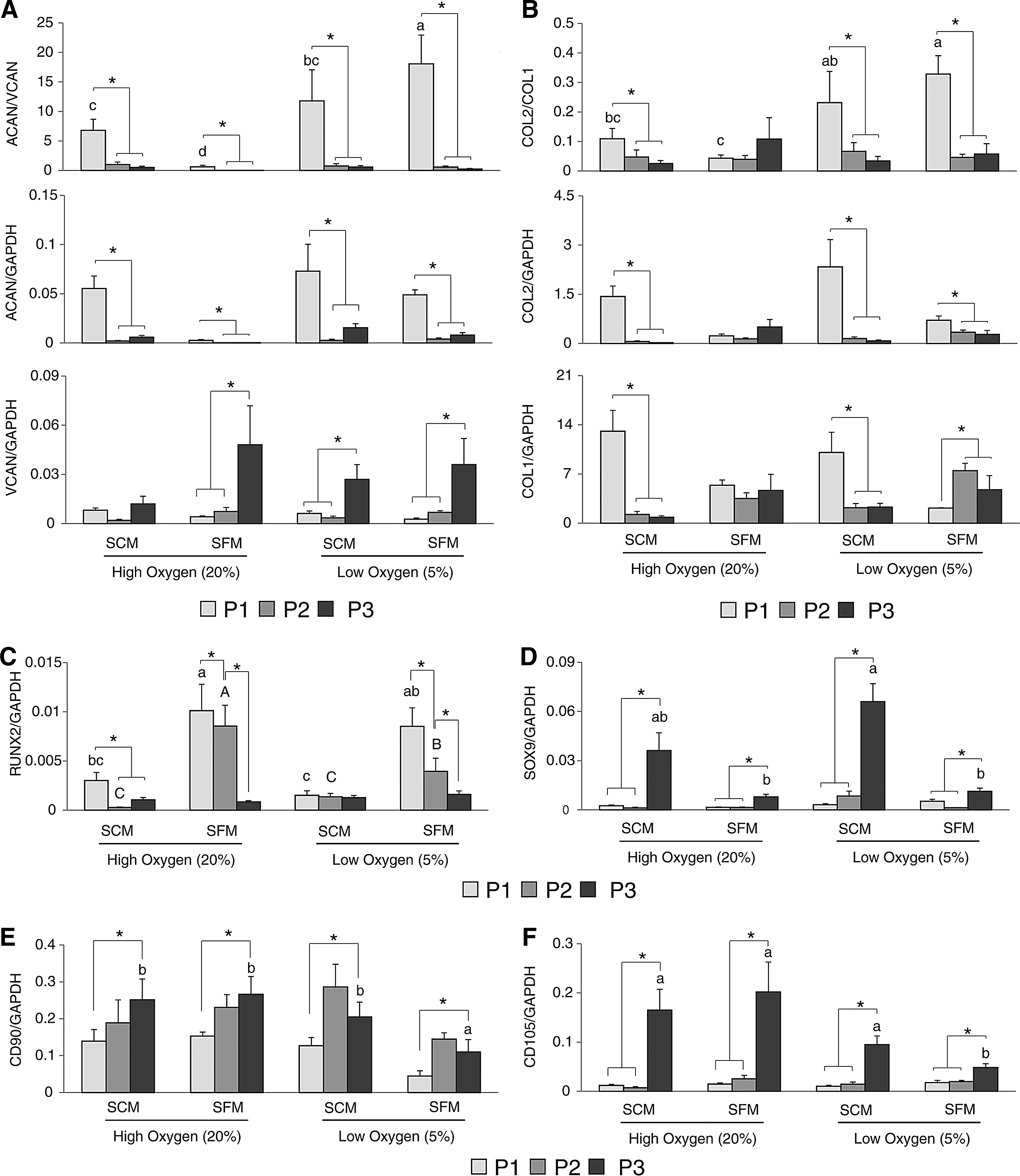
Quantitative gene expression analysis of chondrocytes during in vitro expansion.
SOX9, another transcription factor, demonstrated increased expression with increased passages in all treatments (Fig. 3). Both SCM groups (High-SCM and Low-SCM) had the highest expression of SOX9 at P3 (Fig. 3). CD90 (THY-1) and CD105 (ENG) expression was measured to monitor the de-differentiation status of in vitro cultured chondrocytes. The expression of both CD90 and CD105 increased after three passages in all four treatments (Fig. 3), with CD105 showing a high degree of upregulation in all treatments by P3. Low-SFM demonstrated the lowest expression of both CD90 and CD105 after three passages (Fig. 3).
In vitro re-differentiation of chondrocytes
To evaluate the re-differentiation capacity of in vitro expanded chondrocytes under different conditions, chondrocytes were pelleted and induced to produce ECM. After 6 weeks of induction, the resulting pellets were larger than control pellets that were cultured in incomplete re-differentiation medium (p < 0.05, Fig. 4). The pellets from High-SFM, Low-SCM, and Low-SFM were larger than those from High-SCM. Alcian blue staining revealed the differential production and accumulation of GAGs in the pellets (Fig. 4). Semi-quantitative analysis of the intensity of Alcian blue staining from three different layers (Zone 1, Zone 2, and Zone 3) indicated that Low-SFM cultured chondrocytes produced the most homogenous GAGs, as shown by the consistent intensity of Alcian blue in all layers (Fig. 4). High-SCM, High-SFM, and Low-SCM had a significantly lower concentration of GAGs in the inner layer (Zone 3) (p < 0.05, Fig. 4).
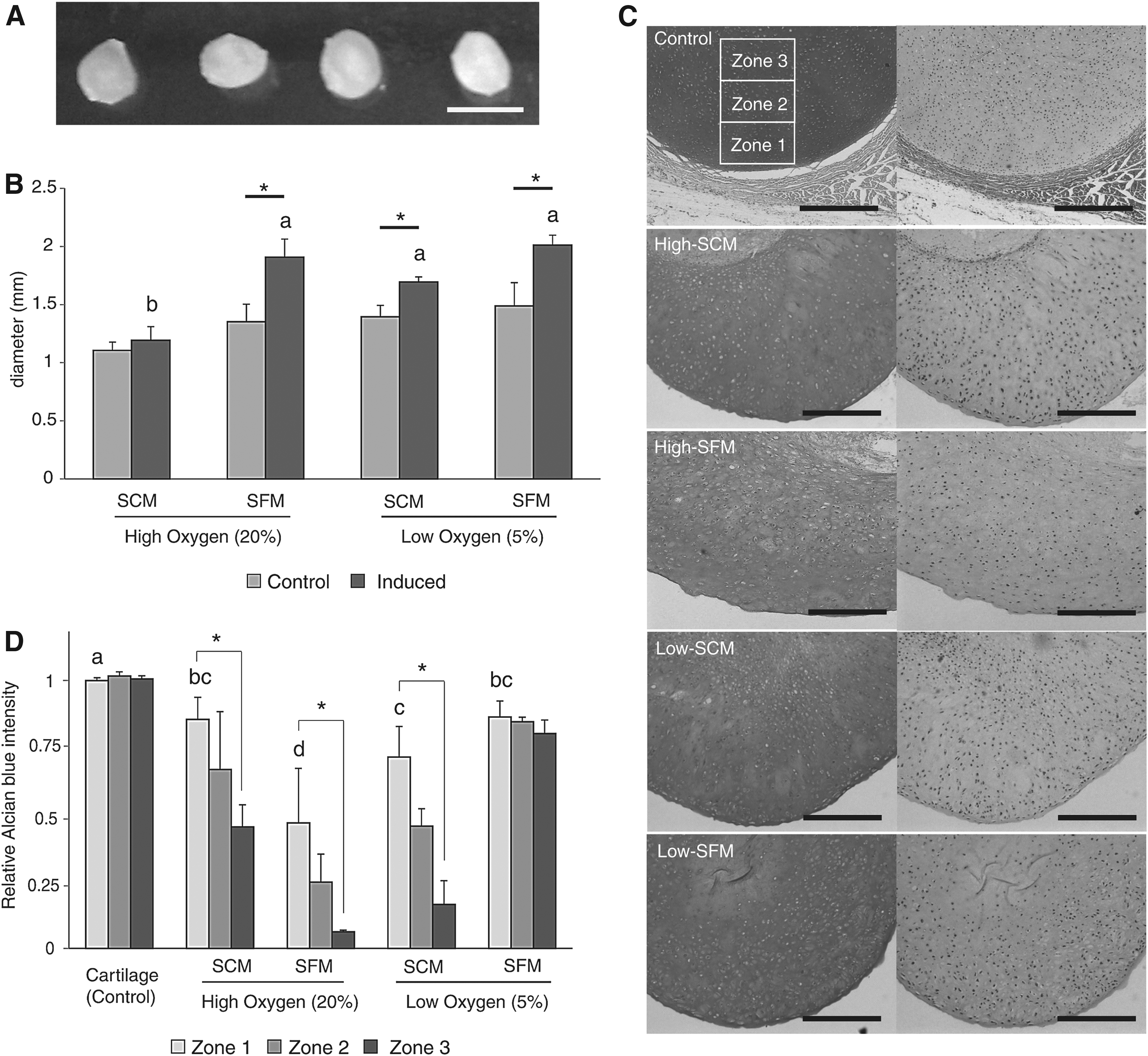
Re-differentiation capacity of in vitro expanded chondrocytes.
Marker expression of re-differentiated chondrocyte pellets
The expression ratio of ACAN/VCAN and COL2/COL1 in the pellets was evaluated by using real-time PCR. The Low-SFM had similar ratios of ACAN/VCAN expression ratio when compared with native cartilage, whereas the Low-SCM had the highest level of ACAN/VCAN ratio (p < 0.05, Fig. 5). Individually, High-SFM exhibited the highest expression of both ACAN and VCAN, resulting in the lowest of the ratios (p < 0.05, Fig. 5). Low-SFM also had the highest expression ratio of COL2/COL1 (p < 0.05, Fig. 5). Both high oxygen groups (High-SCM and High-SFM) had a higher expression of COL1 compared with low oxygen groups (Low-SCM and Low-SFM) and control (p < 0.05, Fig. 5).
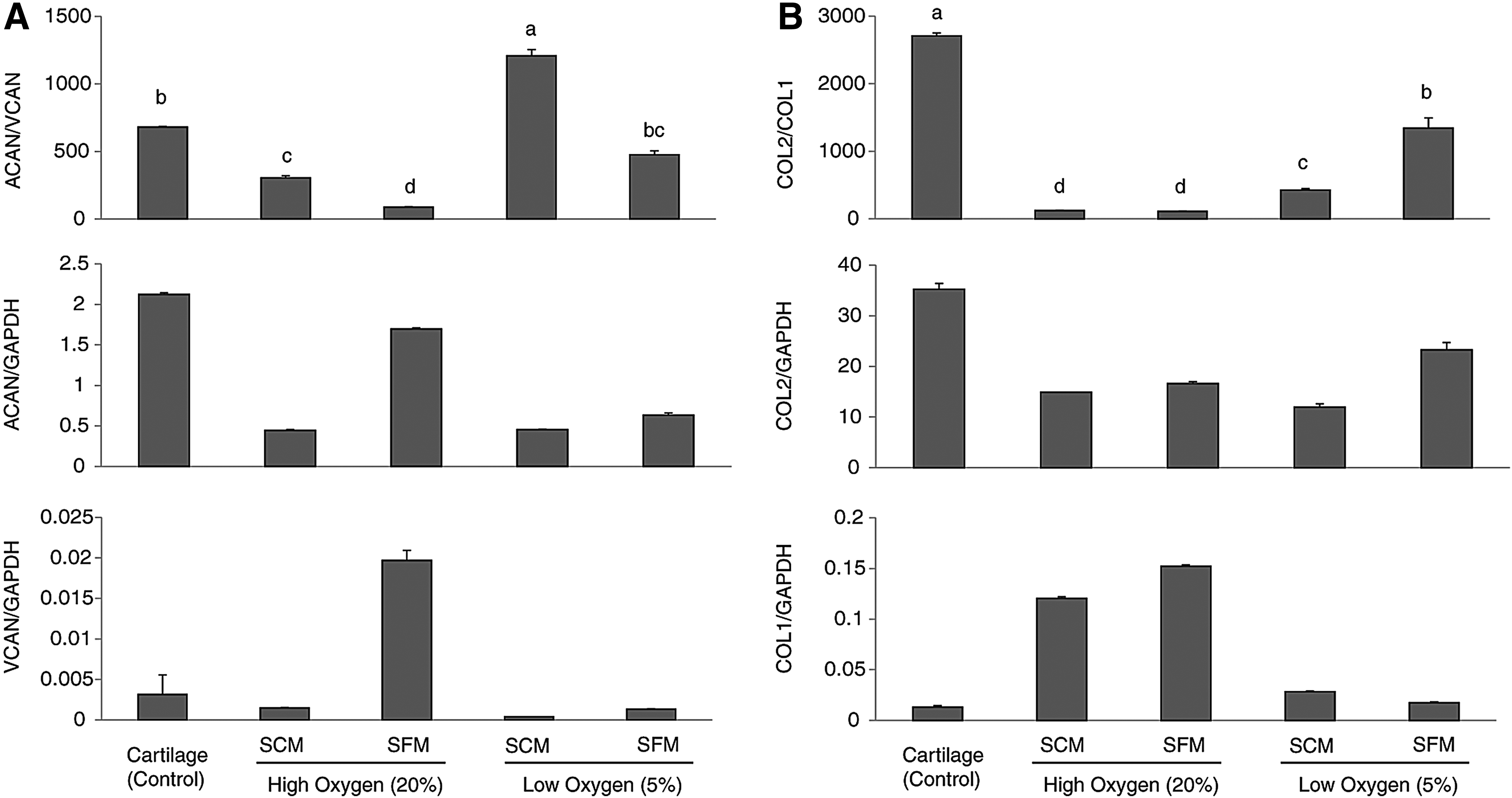
Quantitative gene expression analysis of chondrocyte pellets after 6 weeks of induction.
Quantitative proteome profiles of cultured pellets
Proteomic profiles were obtained from three different native cartilage tissues (ear, joint and rib) and pellets generated from four different cell sources (HF: high-SCM, HS: high-SFM, LF: low-SCM, and LS: low-SFM). Cartilage samples from three different animals were pooled before analysis, and pellets from three separate culture experiments were likewise pooled, resulting in the average of three biological samples for each analysis. A total of 15,544 peptide sequences were obtained and analyzed to identify 515 proteins that belong to 236 protein groups (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/cell).
The relative abundances of each protein between samples were determined compared with a reference sample (articular cartilage). Hierarchical clustering showed that the proteome profile obtained from the pellet composed of chondrocytes expanded in LS had the most similar pattern to native cartilage (Fig. 6). All the identified protein groups in the LS pellets (217) were also expressed in articular (217/236) and costal (202/219) cartilage, and only two protein groups were identified as articular-specific cartilage (Fig. 6 and Supplementary Table S1). The number of identified proteins also varied between each treatment, and only the LS demonstrated a comparable number of proteins to those of native cartilage (Fig. 6).
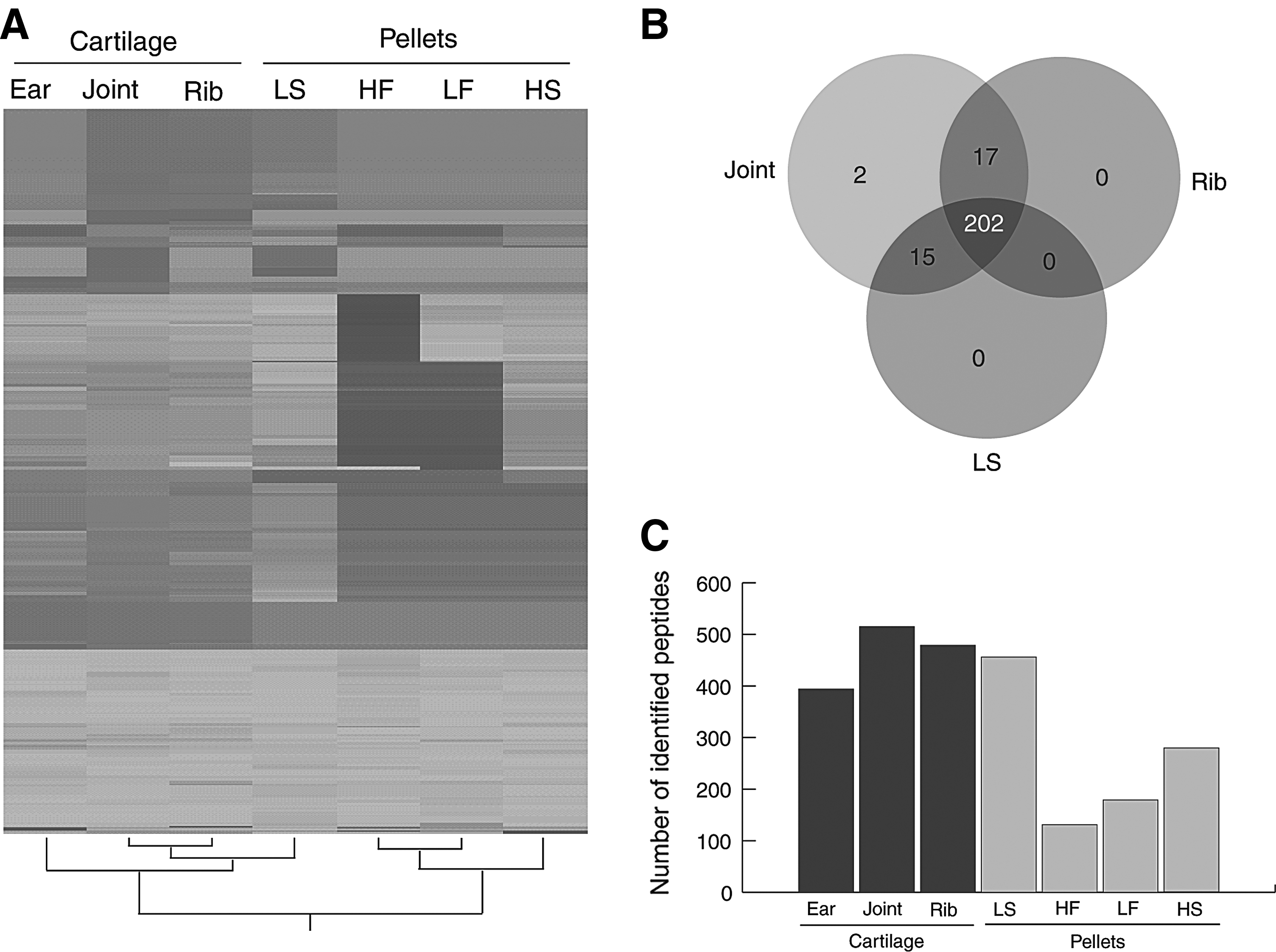
Comparison of proteomic profiles between native and in vitro produced cartilage. The cartilage from three different animals was pooled, as were the pellets from three separate culture experiments, resulting in an average of three biological samples.
Discussion
We report here a novel serum-free culture condition for swine chondrocytes that enables the generation of high numbers of high-quality chondrocytes, a key component to successful cartilage tissue engineering. Our results show that a combined treatment of low oxygen and FGF2 without serum (Low-SFM, LS) during in vitro culture reduces hypertrophic changes of cultured chondrocytes while promoting proliferation (Figs. 1 and 2) and maintaining their ability to produce cartilage matrix (Fig. 4). Further, the chondrocytes cultured in LS condition were able to produce cartilage matrix with a protein composition that more closely resembled native cartilage (Figs. 5 and 6).
Chondrocytes or MSCs are being used for cartilage tissue engineering and/or ACI. For ACI, about 2–5 million cells are needed to cover a 1 cm2 cartilage defect or >240 million cells are required to cover 120 cm2, the average surface area of an entire knee joint (Eckstein et al., 2001). To be able to generate these cell numbers, chondrocytes are isolated and expanded in culture under a variety of conditions such as high-density culture (Watt, 1988), embedding in alginate gels (Benya and Shaffer, 1982) or in three-dimensional cell aggregates (Murphy and Polak, 2004) to promote restoration and maintenance of chondrocyte phenotypic characteristics. However, it has been shown that in vitro chondrocyte expansion induces rapid de-differentiation, reduced proliferation rates, and loss of their re-differentiation capacity (Giovannini et al., 2010b; Mandl et al., 2004b; Yang et al., 2006).
The de-differentiation is accompanied by gradual shifts from Collagen 1 (COL2) to Collagen 2 (COL1) and Aggrecan (ACAN) to Versican (VCAN) expression, resulting in lowered ratios of COL2/COL1 and ACAN/VCAN (Schnabel et al., 2002). As a result, de-differentiated chondrocytes produce COL1-enriched fibrocartilage with less functional biomechanical characteristics compared with native cartilage (Setton et al., 1993). Silencing of COL1A1 by using genetic tools such as siRNA in the presence of cytokines, BMP-2 or TGF-β1 has demonstrated improved cartilage ECM synthesis (Legendre et al., 2013; Perrier-Groult et al., 2013). However, conventional chondrocyte expansion methods include the use of animal serum. Since there are large variations between batches of serum (Yaeger et al., 1997), it becomes difficult to develop a stable, consistent culture system that is amenable to clinical applications and FDA approval.
Thus, it is critical to provide a defined culture environment that promotes chondrocyte proliferation while maintaining their ability to produce functionally relevant cartilage matrix on induction. In addition, the availability of articular chondrocytes for autologous transplantation can be hampered by both the presence of defects in the existing joints and the presence of high levels of inflammation in cases such as osteoarthritis. Costal cartilage, in contrast, is abundant, easily accessible, and has been used extensively in the past for clinical applications related to reconstructive surgery (Lee et al., 2011). Combined with the ability of cells to trans-differentiate if given the proper inductive signaling, costal cartilage could be a useful alternative to both MSCs and articular chondrocytes.
In this study, we extended prior observations on the effect of FGF2 and low oxygen during chondrocyte expansion by using xeno-free, serum-free medium. Both low oxygen and FGF2 promoted chondrocyte proliferation and showed a synergistic effect on growth rate. FGF2 increased cell yield by enhancing proliferation rate (Fig. 1), suppressing terminal differentiation (Fig. 2) and increasing re-differentiation capacity (Fig. 4). In addition, the effects were greater when the cells were cultured in a combination of low oxygen and FGF2 (Low-SFM). Previously, it was shown that both the addition of FGF2 to the conventional serum-containing culture media (Adesida et al., 2006, 2012a; Adesida et al., 2012b; Mandl et al., 2004a, 2004b) and low oxygen tension (Murphy and Polak, 2004) increased cell proliferation and the re-differentiation capacity of chondrocytes.
Similar effects have been shown in other types of stem cells such as bone marrow-derived (Chiou et al., 2006; Handorf and Li, 2011; Robins et al., 2005; Solchaga et al., 2005) or adipose-derived (Wang et al., 2005) MSCs. Also, the advantageous effects of FGF2 were enhanced under low oxygen tension in human meniscal fibrochondrocytes (Adesida et al., 2012a, 2012b). In our study, the AP staining results indicated suppressed hypertrophic terminal differentiation in SFM regardless of oxygen level (Fig. 2).
However, the effect was greater in cells from low oxygen levels, again suggesting the advantageous synergic effect of SFM and low oxygen levels. What was most striking were the differences in the ability of the tested culture conditions to yield a selected targeted cell number of 100 million cells. The Low-SFM media exceeded that target by passage 3, whereas the FBS-containing cultures either did not reach the target number and went into senescence (High-SCM) or took two additional passages to do so (Low-SCM; Fig. 1). This has significant clinical implications as it supports the ability of the Low-SFM to reach a clinically relevant target in a much shorter period and reduced passage number, factors that have both biological and cost implications.
High cell yields must be accompanied with cell quality that is conducive to the generation of high-quality cartilage on induction to be relevant to clinical approaches. Our results demonstrate that chondrocytes cultured in a combined treatment of low oxygen and FGF2 (Low-SCM) maintain higher chondrogenic potential, as shown by gene expression (Figs. 3 and 5) and cartilage production (Fig. 4). In our study, SOX9 expression increased during the in vitro culture in all culture conditions tested. Low-SCM demonstrated the highest expression of SOX9 compared with other culture conditions (Fig. 3).
In contrast to SOX9, COLA2A1 and ACAN decreased during culture. Previous studies have demonstrated that the transcription factors, SOX9 and RUNX2, are master regulatory genes during chondrogenesis (Eames et al., 2004; Lefebvre et al., 2001; Lengner et al., 2005; Yamashita et al., 2009; Zhou et al., 2006). Of particular interest, SOX9, along with L-SOX5 and SOX6, is one of the key transcription factors that regulates chondrogenic differentiation of chondrocytes (Akiyama et al., 2002; Han and Lefebvre, 2008; Lefebvre et al., 2001; Liu and Lefebvre, 2015). Preceding high expression of SOX9 is required and necessary in mesenchymal progenitors to initiate chondrogenesis that is accompanied by downstream expression of L-SOX5 and SOX6.
These three key transcription factors ensure activation of cartilage-associated genes such as COL2A1 and ACAN (Akiyama et al., 2002; Han and Lefebvre, 2008; Liu and Lefebvre, 2015). Hence in our study, the loss of COL2A1 and ACAN during the expansion of chondrocytes indicates that these chondrocytes might have changed their properties (i.e., de-differentiation), but more importantly, high expression of SOX9 suggests that these cells may have higher chondrogenic potential. Indeed, the chondrocytes cultured in Low-SCM that had the highest SOX9 expression among the treatments demonstrated highly improved chondrogenic potential shown by homogenous GAG production (Fig. 4) and a proteomic phenotype similar to naïve cartilages (Fig. 6).
Both SOX9 and RUNX2 are expressed during chondrogenic differentiation of MSCs, and their precisely balanced expression controls proliferation and maturation of chondrocytes. In our studies, the expression of SOX9 and RUNX2 was mainly affected by the presence of serum and cell passage, whereas oxygen level did not have a significant impact on the SOX9 expression (Fig. 3D). The combined results suggest that SCM-induced hypertrophy might be caused by unbalanced expression of SOX9 and RUNX2. De-differentiation of chondrocytes during in vitro expansion was also demonstrated by a decreased expression ratio of COL2/COL1 and ACAN/VCAN and by the increased expression of CD90 and CD105 (Fig. 3).
Both CD90 and CD105 have been reported as markers for de-differentiation of chondrocytes and re-differentiation capacity of de-differentiated chondrocytes as well (Giovannini et al., 2010; Pei and He, 2012). CD90 was found in 99.98% of dedifferentiated articular chondrocytes (Diaz-Romero et al., 2005), and CD90+ cells are also found to positively correlate with chondrogenic potential in MSCs (Nagase et al., 2008). Similarly, it was previously shown that adipose-derived stromal cells (ADSCs) are very analogous to MSCs in their chondrogenic potential and both ADSCs and MSCs contain >90% of CD90-positive cells (Gimble and Guilak, 2003).
Combined with our results, this suggests that though rapid de-differentiation takes place during monolayer expansion of chondrocytes, the de-differentiated chondrocytes population still has the potential to re-differentiate into cartilage matrix-forming cells, and this potential is further enhanced by SFM and low oxygen. This was confirmed by both the Alcian blue analysis of the generated pellets and more importantly, by the proteomic analysis (Figs. 4 and 6).
Notably, the proteomic analysis showed that cells grown in Low-SFM media produced cartilage that most closely resembled native cartilage. This was dramatically demonstrated by the clustering analysis that showed that the only in vitro produced cartilage that clustered with the in vivo native cartilage was the one generated from Low-SFM (Fig. 6).
More interestingly, even though the initial cells were derived from costal cartilage, the proteomic analysis clustered the Low-SFM sample with both articular (joint) and costal (rib) cartilage (Fig. 6). This again supports costal cartilage as an alternative source for cartilage tissue engineering. Conventionally, the quality of cartilage is determined by the use of Western blot and the detection of a few key proteins such as type II collagen.
This approach, however, results in a very superficial description of the protein composition of cartilage generated under different conditions. When compared with Western-blot, MS is more comprehensive, robust, quantitative, and rigorous (Aebersold et al., 2013). In addition, several groups have reported the ability to trans-differentiate the cartilage of one type to another by co-culture methods. This has been accomplished not only between different types of chondrocytes (Kuhne et al., 2010) but also between chondrocytes and other stem cells, including MSCs (Bian et al., 2011; Dai et al., 2012). The combined results support the fact that it will be feasible to design systems that allow rapid expansion of costal chondrocytes combined with induction protocols that shift cartilage production toward articular cartilage.
In addition, by utilizing genetically modified pigs designed for xenotransplanation (Nieman and Petersen, 2016), it should be feasible to utilize porcine-derived chondrocytes and the culture conditions described here to develop large numbers of cell that can be used for the treatment of cartilage-associated disorders in humans.
Taken together, our study showed that the combined application of defined serum-free media and low oxygen provides an optimized chondrocyte expansion method that yields high-quality cartilage on induction. The optimized culture conditions enhanced cell yield, as well as lowered de-differentiation and hypertrophic differentiation. Further, Low-SFM cultured chondrocytes produced cartilage ECM that was the most similar to native cartilage after induced re-differentiation. The combined cell culture strategy of low oxygen level and SFM will improve ACI and cartilage tissue engineering by providing a high yield of high-quality chondrocytes, a potentially superior alternative to MSCs and articular cartilage.
In addition, the availability of a large animal model will allow us to test the utility of the developed system for tissue repair and regeneration, and this, in turn, will facilitate the development of protocols that can be rapidly moved into a human clinical setting. The use of gene-edited pigs to be used for xenotransplantation would further expand the utility of this work (Nieman and Petersen, 2016).
Footnotes
Acknowledgments
Funding was provided by the Comparative Medicine Institute (CMI). The authors are grateful to Dr. Elizabeth Loboa for editing and content suggestions that considerably improved this article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.