
Abstract
Abstract
Specific activation of endogenous genes can be achieved by programmable artificial transcription factors (ATFs). In this study, we compared two artificial, programmable, clustered regularly interspaced short palindromic repeats (CRISPR)-based, ubiquitous transcription factors: deficient CRISPR-associated protein 9 (dCas9)-VP64 (CRISPRa) alone, or a combination of dCas9-VP64 and MS2-P65-HSF1 [synergistic activation mediator (SAM) system] mediated activation of five pluripotency genes: KLF4 (K), LIN28 (L), MYC (M), OCT4 (O), and SOX2 (S) in human cells (HEK293T, HeLa, HepG2, and primary fibroblasts). Activation potential was monitored using a luciferase reporter system and we found that both CRISPRa and SAM can efficiently activate the proximal promoter of all five genes. We also observed that the guide RNA (gRNA) target sites and number of gRNAs have a major effect on gRNA-guided activation efficiency. Furthermore, increased activation efficiency (>3-folds) could be achieved by the SAM system compared to CRISPRa. In addition, we discovered that only the SAM system could efficiently activate LIN28, OCT4, and SOX2 expression (up to 100-folds compared to coexpression with a scrambled gRNA) in primary human fibroblasts. This SAM-mediated activation of LOS can be stably maintained for over 20 days in fibroblasts cultured in either fibroblasts or stem cell medium. However, when attempting to use the SAM-LOS activation as an approach for induced pluripotent stem cells-reprogramming, no embryonic stem-like colonies could be obtained from these SAM fibroblasts. In conclusion, our study showed that CRISPR/Cas9-based ATFs are potent to activate and maintain transcription of endogenous human pluripotent genes. However, future improvements of the system are still required to improve activation efficiency and cellular reprogramming using ATFs.
Introduction
E
An alternative approach would be the direct activation of the endogenous pluripotency genes instead of delivering the reprogramming factors exogenously. Previous studies have reported that human endogenous pluripotency genes, including OCT4, SOX2, MYC, and KLF4, can be activated by artificial transcription factors (ATFs) via zinc fingers (Ji et al., 2014) or transcription activator-like effectors (TALEs) technology (Chavez et al., 2015; Gao et al., 2013; Hu et al., 2014). However, the broader application of these technologies is limited by the complexity and uncertainty of programmable vector generation and unpredicted targeting efficiency.
Recently, the clustered regularly interspaced short palindromic repeats (CRISPR) and nuclease-deficient CRISPR-associated protein 9 (dCas9)-based systems have been applied for ATFs construction and regulation of gene expression (Gilbert et al., 2013; Qi et al., 2013). By fusing dCas9 to a transcription effector factor domain such as VP64, the dCas9-VP64 ATF can specifically bind to any promoter region in cells, guided by a small guide RNA (gRNA). This specificity is defined by the complementary guide sequences in the gRNA and the target site. To enhance the activation efficiency, Konermann et al. (2015) fused a dimerized MS2 bacteriophage coat protein binding hairpin aptamer to the gRNA scaffold.
The modified system, called synergistic activation mediator (SAM), comprises MS2-gRNA, dCas9–VP64, and the MS2–p65–HSF1 fusion protein. The MS2–p65–HSF1 fusion protein contains MS2 bacteriophage coat protein fused to the NF-kappa-B (P65) and heat shock transcription factor 1 (HSF1) transcription factors (hereafter) (Konermann et al., 2015). The CRISPR/Cas9 activation strategy has provided an open-resource and powerful tool for targeting human reprogramming factors including KLF4, LIN28, MYC, OCT4, NANOG, and SOX2 in various human cell types (Cheng et al., 2013, 2016; Mali et al., 2013; Perez-Pinera et al., 2013; Hu et al., 2014; Ji et al., 2014; Balboa et al., 2015). However, no research so far has reported successful reprogramming and production of iPSCs by the CRISPR/Cas9 activation system. This might stem from the fact that until now it has not been sufficiently investigated how long pluripotency genes are able to retain their activity after activation through the CRISPR/Cas9-based activation system and whether this kind of ATF could facilitate bona fide reprogramming of somatic cells to iPSC.
In this study, we investigated the ability of the CRISPR/dCas9-VP64 and CRISPR SAM activation systems to activate OCT4, SOX2, MYC, LIN28, and KLF4 gene expression in various human cell types and tested the potential use of the SAM system to induce pluripotency in somatic cells.
Materials and Methods
Cells and culture condition
HEK293T, HeLa, and HepG2 cells (original purchased from ATCC), and primary normal human dermal fibroblasts (anonymous), were cultured in Dulbecco's Modified Eagle's medium (DMEM) (high glucose), supplemented with 10% FCS, 1% penicillin-streptomycin, and 1% glutamax and incubated at 37°C with 5% CO2. SAM fibroblasts, which expressed dCas9-VP64 and MS2-P65-HSF1, were maintained in the culture medium supplemented with 1 μg/mL blasticidin and 100 μg/mL hygromycin.
gRNA design and construction
All gRNAs were designed using the online optimized CRISPR design tool (http://crispr.mit.edu) and targeted the proximal promoter regions of KLF4, LIN28, MYC, OCT4, and SOX2 (listed in Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/cell). Oligos, synthesized by Eurofinsgenomics, were annealed and sub-cloned into the lentiGuide-puro vector (a gift from Feng Zhang's lab, Addgene plasmid no. 52963) or the lenti-single guide RNA (sgRNA) (MS2)-zeo vector (a gift from Feng Zhang's lab, Addgene plasmid no. 61427).
Firefly luciferase reporter plasmid construction and activity assay
The proximal promoter region of KLF4, LIN28, MYC, OCT4, and SOX2 was amplified from human genomic DNA using primers shown in Supplementary Table S2. KpnI and HindIII restriction enzyme recognition sites were included via the forward and reverse primer, respectively (Supplementary Table S2). The polymerase chain reaction (PCR) products were cloned into a pGL3 firefly luciferase reporter plasmid (Promega, San Luis Obispo, CA) via KpnI and HindIII-based cloning (Fig. 1A). Sanger sequencing and restriction enzyme digestion validated successfully generated reporter vectors.
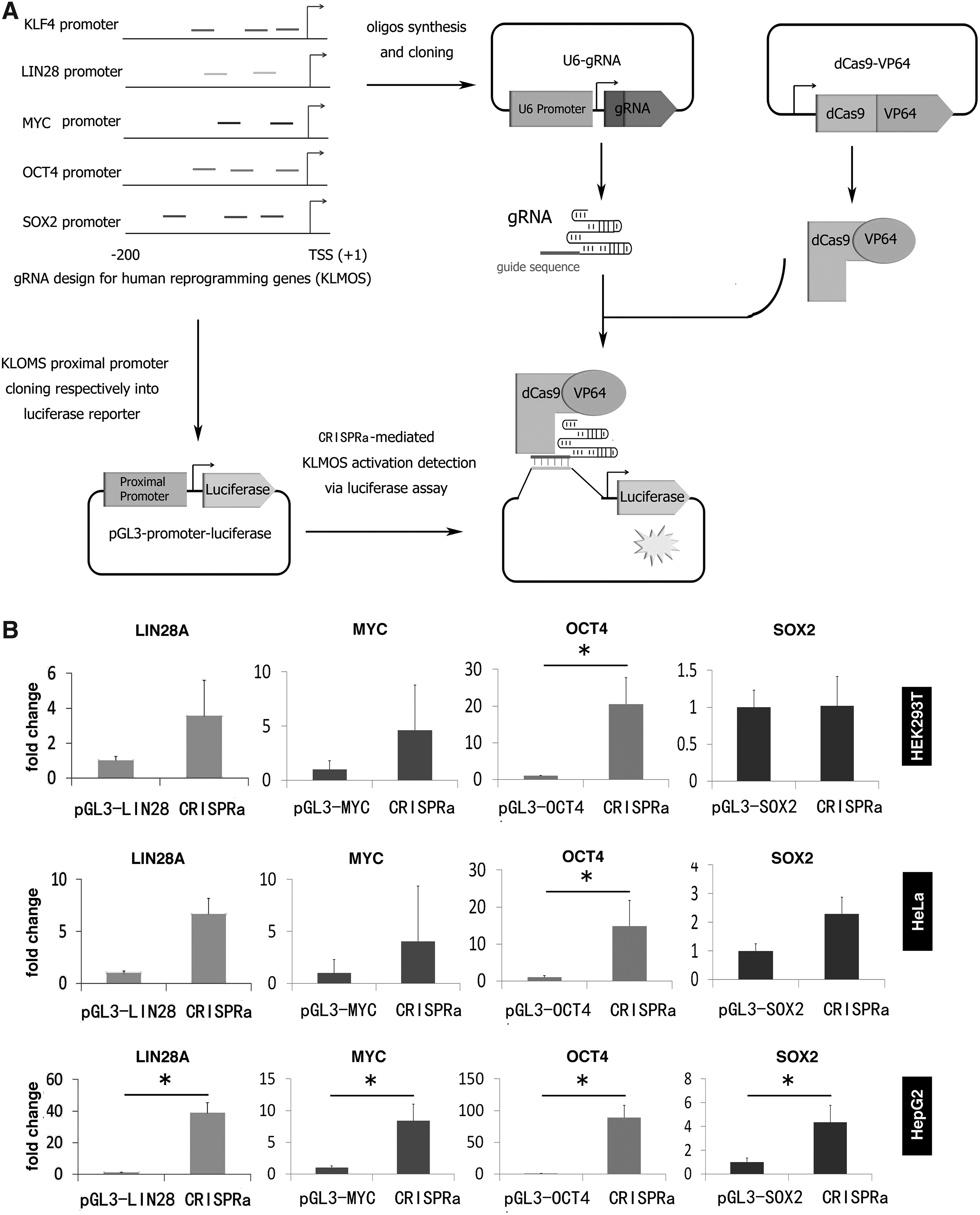
Luciferase reporter activation induced by CRISPRa.
Luciferase assays were carried out according to manufacturers' instructions of the Dual-Glo® Luciferase Assay System (Promega, E2940). Briefly, 10,000 cells were seeded per well in 96-well white plates 24 hours before transfection. Transfections were performed using the X-tremeGene 9 (Roche Life Science) kit according to the manufacturer's protocol. Each transfection master mix (5X) contained plasmids of gRNAs (total 100 ng), dCas9-VP64 (100 ng), firefly luciferase reporter vector (150 ng), control renilla luciferase expression vector (50 ng, Promega), and either 100 ng MS2-P65-HSF1 (for SAM system) or 100 ng loading control plasmid pUC19 (for CRISPRa system). Cells transfected only with the luciferase promoter reporter vector at the same amount like for the experiments were used as controls for each gene activation analysis. The transfection master mix was added into 5 wells in a 96-well plate (representing 5 experiment repeats). Cells were subjected to luciferase assays 48 hours after transfection using the Dual-Glo Luciferase Assay System.
Lentivirus production
HEK293T cells (Life Technologies) were cultured in DMEM (Gibco Technologies) supplemented with 10% fetal bovine serum (FBS) (Seradigm), 1% Glutamax (Life Technologies), and 1% penicillin/streptomycin (Life Technologies). One day before transfection, 1 × 107 cells were seeded on a 15 cm plate with 15 mL medium. Cells were transfected the next day at 80%–90% confluence. For transfection, 31.5 μg of plasmid containing the vector of interest, 31.5 μg of pMDGP-Lg/pRRE, 9.07 μg of pMD2.G and 7.26 μg of pRSV-REV (lentivirus packaging plasmids were generously provided by Prof. Jacob Giehm Mikkelsen from the Department of Biomedicine, Aarhus University) were diluted in 1089 μL milliQ water and mixed with 121 μL of 2.5 M calcium chloride solution and 1210 μL of 2 × HBS solution. After 20 minutes incubation at room temperature (RT), the solution mixture was added directly to the cells. Twenty-four hours after transfection the medium was changed. Virus supernatant was harvested 48 and 72 hours after transfection, filtered with a 0.45-mm polyvinylidene difluoride (PVDF) filter (Millipore), and stored at −80°C.
Lentivirus transduction
Normal human dermal fibroblast cells (anonymous) were cultured in DMEM (Gibco Technologies) supplemented with 10% FBS (Seradigm), 1% Glutamax (Life Techonologies), and 1% penicillin/streptomycin (Life Technologies) and passaged at a 1:3 ratio when cells reached 70%–80% confluence. For generation of the SAM fibroblasts, cells were transduced with crude lentivirus in a T75 flask. About 7.5 × 105 cells were plated onto the T75 flask 1 day before virus transduction. Fifteen microliter of crude virus with 8 μg/mL polybrene (Sigma) were added to the flask. The lentiviral supernatant was replaced with fresh medium 48 hours post-transduction. Selection agents were added immediately after the medium change [blasticidin (2 μg/mL) and hygromycin (200 μg/mL), Life Technologies].
The blasticidin and hygromycin concentration was optimized by a kill curve assay in human fibroblasts. Medium was replaced every other day during the selection period of 7 days. Surviving cells were passaged and cultured in medium with 1 μg/mL blasticidin and 100 μg/mL hygromycin. For transduction of SAM fibroblasts with gRNA expressing lentivirus, 1 day before plating, SAM fibroblasts cells were detached with trypsin and reseeded on 24-well plates for transduction at a density of 2 × 104 cells per well. Each well of cells was infected by 1 mL lentiviral supernatant. Cells were either harvested for DNA or RNA isolation at 72 hours after transduction. To titer the lentiviral prep, we transduced SAM fibroblasts using a batch of green fluorescent protein (GFP)-expressing lentiviral particles. The GFP particles were generated in the same experiment while producing gRNA expression lentivirus particles. A multiplicity of infection (MOI) of 0.9, which correspond to 90% transduction, was chosen for the gRNAs (1 mL crude virus, Supplementary Fig. S1).
Quantitative PCR analysis
RNA samples were isolated using RNeasy Mini Kits (Qiagen) following the manufacturer's instructions. The complementary DNA (cDNA) was generated using iScript cDNA Synthesis Kit (Bio-Rad) (Promega) and quantitative PCRs (qPCRs) were performed using SYBR Green I Master Kit (Roche) with primers listed in Supplementary Table S3. Relative gene expression level in fold change was calculated by the 2−ΔΔCt method: Target threshold cycle (Ct) values were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Ct values, and fold changes in target gene expression were determined by comparing to experimental controls expressing a scrambled gRNA. qPCR analysis was performed in triplicates of technical repeats for each sample. RNA isolated from iPSC controls, grown in E8 medium with a feeder-free culturing system, were used as a positive control.
OCT4 promoter methylation analysis by bisulfite pyrosequencing
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, 69506) according to the manufacturer's instructions. Genomic DNA (100 ng) was bisulfite treated using the EpiTect Bisulfite Kit (Qiagen, 59104) according to the manufacturer's protocol to convert unmethylated cytosines to uracils. The bisulfite converted DNA was used for PCR amplification of the OCT4 target. PCR was performed in a 25 μL volume containing 0.15 μL Hotstar Taq polymerase (5 U/μL) (New England Biolabs, M0495L), 2.5 μL 10x Standard buffer, 0.5 μL of 10 mM deoxynucleotides (dNTPs), 1.0 μL of each primer (10 μM), and 1.5 μL bisulfite converted genomic DNA. PCR was performed under the following conditions: 95°C for 5 minutes followed by 45 cycles of 94°C for 30 seconds, 58°C for 1 minutes, and 72°C for 45 seconds, and, finally, by 72°C for 7 minutes. Four microliter PCR product was checked by gel electrophoresis. Pyrosequencing was performed with the PyroMark Q24Advanced Reagents (Qiagen, 970922) using 20 μL bisulfite PCR product and 20 μL sequencing primer (0.375 μM) according to the PyroMark Q24 CpG protocol quantifying the percentage of methylated CpGs.
Flow cytometry analysis
Seventy-two hours after transduction, cells of each well were harvested, washed once in phosphate-buffered saline (PBS), and resuspended in 400 μL of 4% paraformaldehyde followed by a 10 minutes RT incubation. The suspension was subsequently centrifuged and washed in PBS-2% FCS. Cells were pelleted and resuspended in cold methanol followed by incubation for 10 minutes at RT, centrifuged and washed twice with PBS-2% FCS, and followed by resuspension in PBS-2% FCS. Approximately 50,000 cells in 100 μL PBS-2% FCS were incubated with conjugated antibody against human OCT4 (PerCP-Cy™ 5.5 Mouse anti-Oct3/4; BD Pharmingen™; Cat. no. 560794; 20 μL per test), human SOX2 (Alexa Fluor® 647 Mouse Anti-Sox2; BD Pharmingen; Cat. no. 562139; 5 μL per test), and human LIN28 (Alexa Fluor 488 Mouse anti-Human LIN28; Cat. no. 563597; 5 μL per test) for 1 hour at 37°C, washed twice in PBS-2% FCS, and resuspended in PBS. Cells were analyzed using LSRFortessa analyzer (BD Bioscience). For each group, 20,000 cells were collected and subsequently analyzed using FlowJo software.
Statistics
Unless stated elsewhere, all data in this study were presented as mean ± 1 SD (n = 3, triplicates in independent transfection or transduction). A p < 0.05 was considered statistically significant by analysis of variance (ANOVA) or t-test analysis.
Results
Evaluation of CRISPRa-mediated activation of pluripotency genes using a luciferase reporter assay
To investigate the activation of four different pluripotency genes LIN28A, MYC, OCT4, and SOX2 (these four genes hereafter abbreviated as LMOS) by the CRISPR/dCas9-VP64 system (CRISPRa), we constructed a firefly luciferase reporter system, in which the proximal promoter of LIN28A, MYC, OCT4, or SOX2 was cloned into a firefly reporter vector (Fig. 1A). We also designed 2 to 3 gRNAs targeting the proximal promoter of each gene. To select an optimal cell model for CRISPRa-mediated activation, three human cell lines were tested: HEK293T, HeLa, and HepG2. We cotransfected the cell line with each firefly luciferase reporter construct and the dCas9-VP64 expression vector, the corresponding gRNA expression vectors, and a renilla luciferase expression vector (for normalization). Luciferase expression was quantified 48 hours after transfection. We observed that CRISPRa induced significant activation (p < 0.05) of LIN28A, MYC, OCT4, and SOX2 promoters in HepG2 cell lines, but less prominent in HEK293T and HeLa cells (Fig. 1B). Thus, the HepG2 cells were selected for further evaluations of the CRISPRa system.
The effect of gRNA binding site, dose, and multiplexity on gene activation
We next compared the efficacy between CRISPRa and the SAM system (Konermann et al., 2015) using the reporter system in HepG2 cells. The SAM system combines both dCas9-VP64 and MS2-P65-HSF1 in one single CRISPR activation system, which has previously been shown to be efficient for gene activation (Fig. 2A). We transfected each reporter vector alone, or together with either the CRISPRa or the SAM system, into HepG2 cells and analyzed firefly luciferase expression 48 hours after transfection. Consistent with the previous report (Konermann et al., 2015), the SAM system significantly achieved about three to fourfolds higher activation efficiency over the CRISPRa for all luciferase reporter vectors tested (Fig. 2B). Using the OCT4 reporter vector, we also confirmed that expression of dCas9-VP64 and MS2-P65-HSF1 effector alone or together showed no sign of activation, indicating that the activation is mediated by the gRNAs (Fig. 2C).
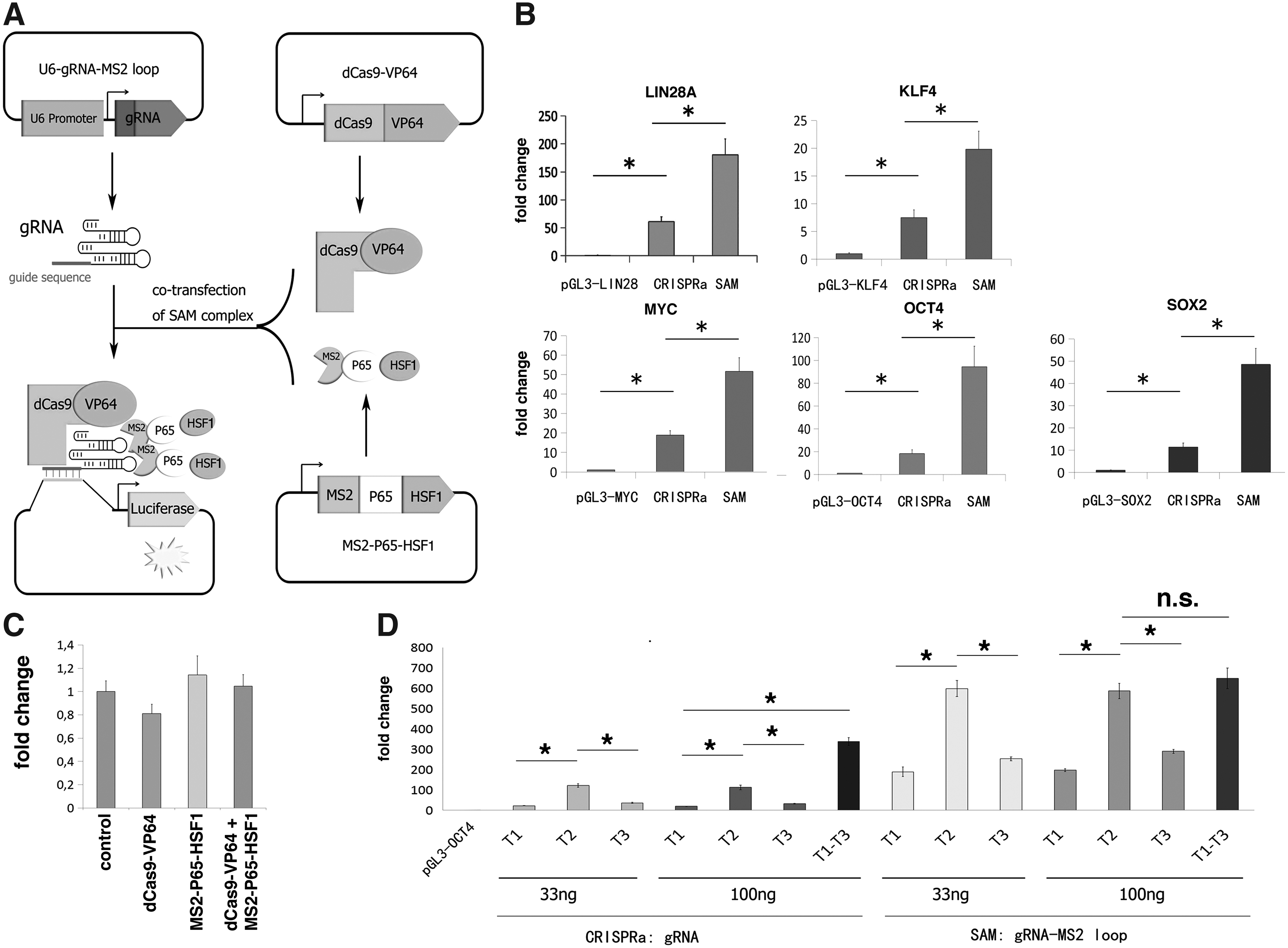
Dose-dependent luciferase reporter activation induced by the SAM system.
To investigate the effect of individual gRNAs, gRNA amount and multiplexity on gene activation, we selected the OCT4 luciferase reporter. We transfected the HepG2 cells with the OCT4 luciferase reporter vector alone or together with the CRISPRa or SAM system. We tested two gRNA amounts (33 or 100 ng). For the usage of multiple gRNAs, a mixture of all three OCT4 gRNAs (33 ng each) was used. As shown in Figure 2D, the OCT4 gRNA Targeting site 2 (hereafter, abbreviated as T2) resulted in significantly higher activation than T1 and T3 (p < 0.05, ANOVA). We also observed that increasing the amount of gRNA (from 33 to 100 ng) did not significantly increase the activation effect, neither in the CRISPRa nor the SAM system. The combination of multiple gRNAs significantly enhanced the activation efficiency of the CRISPRa system compared to sgRNAs (fold change = 3–17, p < 0.05) (Fig. 2D), whereas, such an effect was not observed for the SAM system. The use of OCT4 gRNA T2 alone had resulted in ∼600-fold activation, which might already represent the maximum expression capacity of the proximal OCT4 promoter. Nevertheless, as no adverse effects were noted, a mixture of two to three gRNAs targeting each gene was implemented in our following experiments.
SAM induced transient expression activation of pluripotency genes in human fibroblast
To investigate whether the SAM system can activate the endogenous pluripotency genes in primary fibroblasts, we first established a fibroblast cell line stably expressing dCas9-VP64 and MS2-P65-HSF1. We selected a primary human dermal fibroblast that had previously been tested in iPSC generation (Kang et al., 2015). The fibroblasts were transduced with lentivirus expressing dCas9-VP64 and MS2-P65-HSF1 and cultured in selection medium containing blasticidin and hygromycin for 7 days (Fig. 3A). The SAM fibroblasts were then transduced with lentiviral particles carrying a mixture of individual KLF4, LIN28, MYC, OCT4, and SOX2 gRNA expression cassettes (the transfection group hereafter denoted as SAM-KLMOS) or only the OCT4 gRNA expression cassettes (SAM-OCT4) (Fig. 3A). For positive controls, the SAM fibroblasts were transduced with lentivirus expressing a GFP, a scrambled gRNA, and lentivirus expressing the four reprogramming factors KMOS (KMOS-OE). Three days after lentiviral transduction, we analyzed the expression of endogenous KLMOS genes by qPCR. We noted that these qPCRs are specific for the human endogenous KLMOS, and do not detect the exogenous transcripts from KMOS-OE.
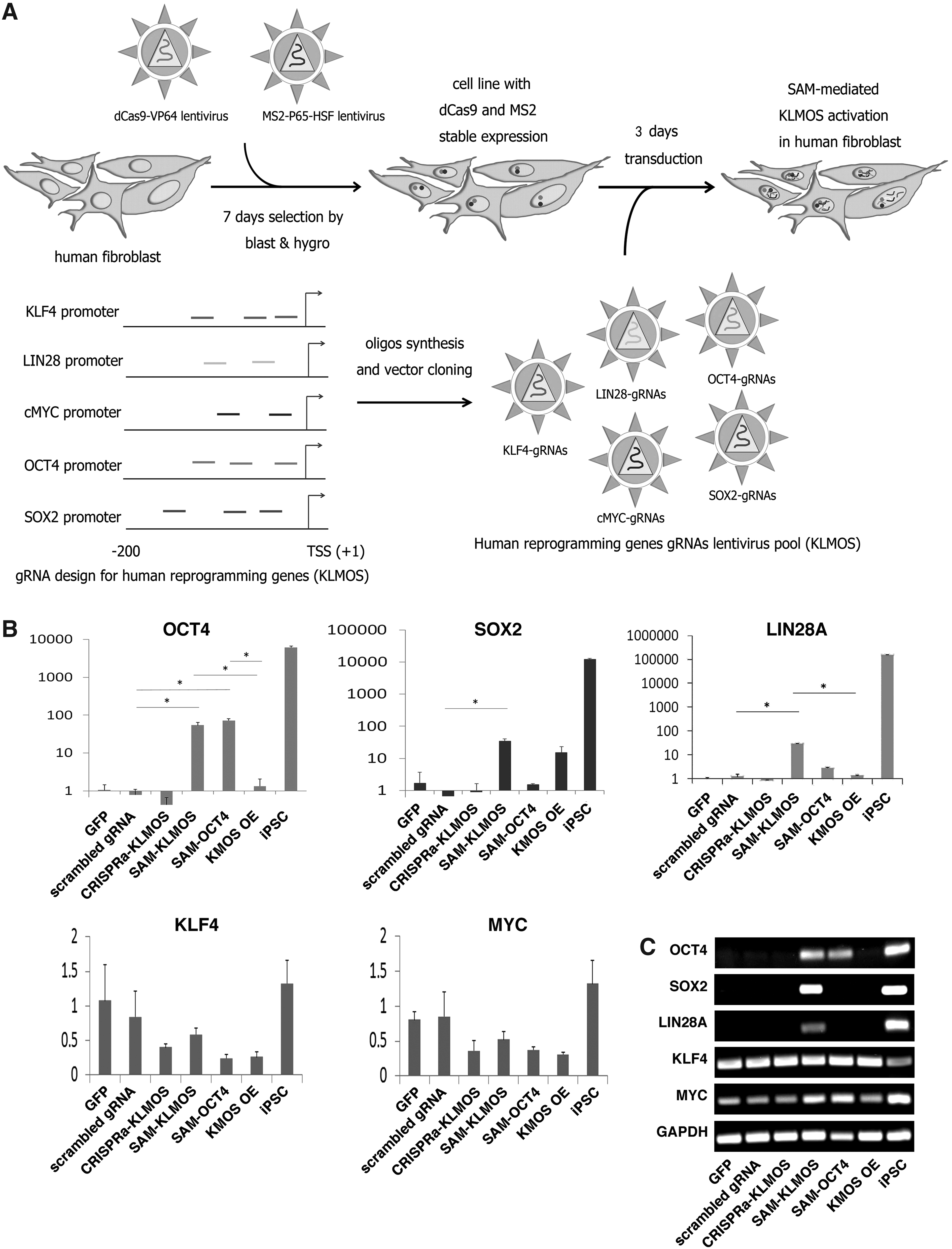
SAM-mediated KLMOS activation in human Fbs.
Our results showed that both SAM-KLMOS and SAM-OCT4 resulted in over 60-fold activation of OCT4 expression (Fig. 3B). SOX2 and LIN28 were also activated by SAM-KLMOS (34- and 29-fold, respectively). In contrast, ectopic overexpression of the four reprogramming factors KMOS (KMOS-OE) only activated the endogenous SOX2 expression ∼10-fold. Neither expression of scrambled gRNA nor GFP led to an increase in KLMOS expression levels (Fig. 3B). The SAM-induced activation of OCT4, SOX2, and LIN28 expression was also confirmed by gel electrophoresis of qPCR products (Fig. 3C). Unlike the SAM system, the CRISPRa gRNAs (CRISPRa-KLMOS), which did not contain the MS2 loop in the gRNA scaffold, failed to activate the endogenous OCT4, SOX2, and LIN28 expression in these SAM fibroblasts (Fig. 3B–C). However, compared to iPSCs, the levels of OCT4, SOX2, and LIN28 activation by SAM were still ∼100-fold lower (Fig. 3B). We did not observe any increase in activation of KLF4 and MYC, since these two genes are already highly expressed in fibroblasts (Fig. 3B, C).
Activation of endogenous OCT4 expression by SAM was not correlated with promoter demethylation
To study whether the SAM-induced activation of OCT4 expression was correlated with a change of methylation status, we tested the DNA methylation status of 7 CpG loci in the OCT4 proximal promoter by bisulfite pyro-sequencing. Neither SAM-OCT4 nor SAM-KLMOS resulted in changes of DNA methylation at the OCT4 promoter investigated (Fig. 4). We did not observe any difference in OCT4 promoter methylation in SAM-OCT4, SAM-KLMOS transduced fibroblasts, or the control fibroblasts transfected with a scrambled gRNA, KMOS overexpression, or GFP. However, this OCT4 promoter region was hypomethylated in iPSCs. These results suggest that SAM-mediated activation of OCT4 expression is not correlated with a change in the promoter DNA methylation status, at least for those CpGs investigated in this assay.
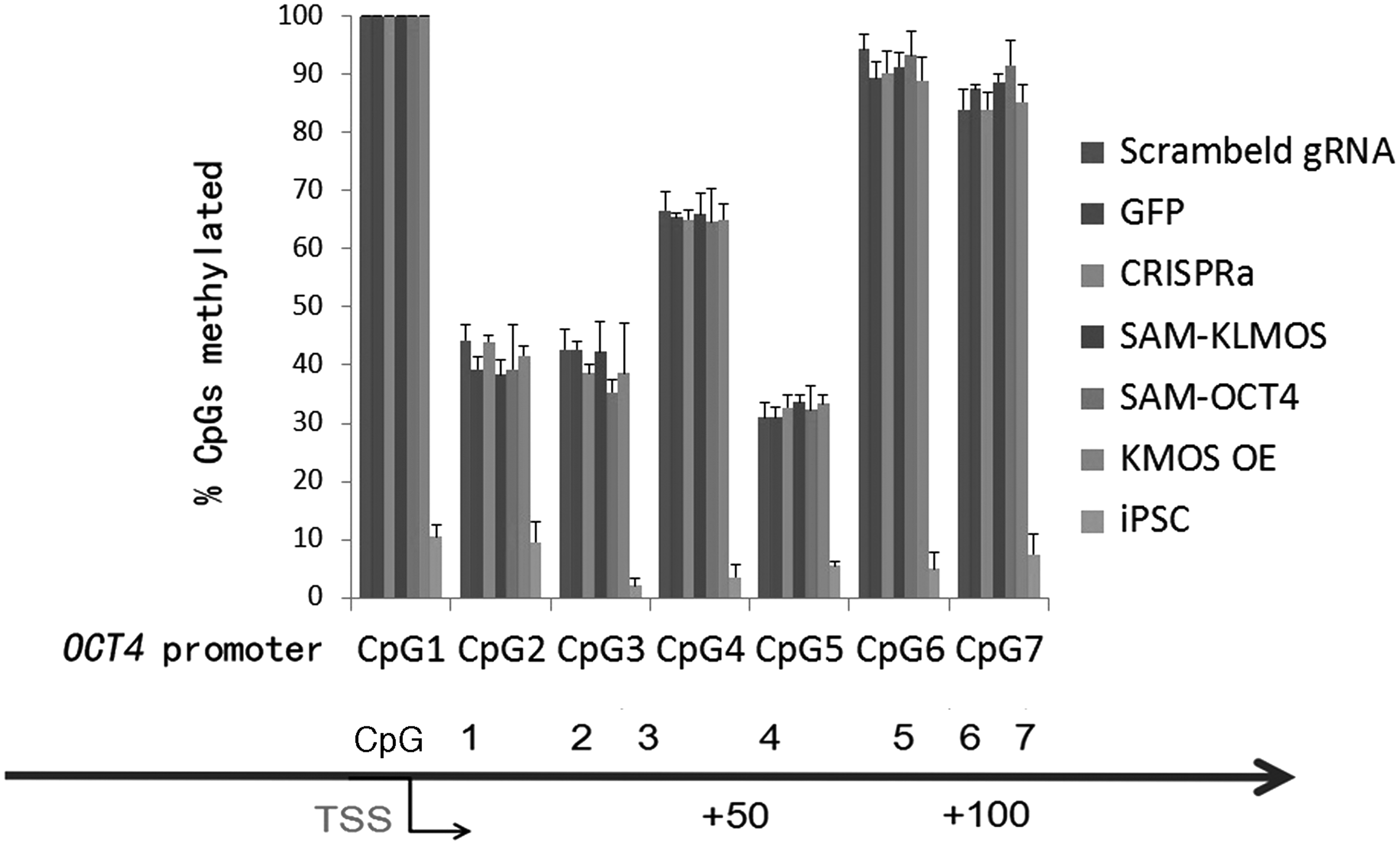
OCT4 promoter methylation analysis. Quantitative bisulfite pyrosequencing in DNA samples isolated from SAM-Fbs at 3 days after transduction. The percentage of methylated CpGs at eight CpG sites of the OCT4 promoter was analyzed, and presented as mean and one SD (n = 3 independent transductions).
SAM-OCT4 induced long-lasting activation of OCT4 expression in human fibroblasts
To address whether the SAM system might impose a protracted effect on gene activation, we analyzed OCT4 expression over 12 days in the SAM-OCT4 fibroblasts in fibroblast medium. As shown in Figure 5A, OCT4 expression was maintained at a high level and over the 12 days post-transduction (more than 100-fold), suggesting that SAM can induce long-lasting activation of OCT4 expression.

Long-term activation of endogenous pluripotent genes expression induced by SAM activation.
SAM-KLMOS was not able to reprogram fibroblasts to iPSCs
To investigate whether the SAM-KLMOS activation could reprogram the fibroblasts to iPSCs, the SAM-KLMOS fibroblasts were seeded on mouse embryonic fibroblast (MEF) feeder 4 days after KLMOS gRNA transduction and maintained in knockout iPSC medium. However, no embryonic stem (ES)-like colonies were observed in the SAM-KLMOS fibroblasts 20 days after plating on the MEF feeders, while in the group of cells transduced with KMOS expression vectors ES-like colonies emerged after 12–14 days. Two iPSC clones expressing dCas9-VP64 and MS2-P65-HSF1 have been generated and presented as public stem cell resource (Xiong et al., 2016). To investigate whether this is due to gene silencing, we measured the expression of all five genes by qPCR in the SAM-KLMOS cells cultured on feeders in iPSC medium for 20 days. We observed that OCT4, LIN28, and SOX2 were still highly expressed in the SAM-KLMOS fibroblasts compared to the SAM fibroblasts transduced with a scrambled gRNA (Fig. 5B).
However, the expression levels were still much lower than those expressed in iPSCs. These expression results were further validated by gel electrophoresis analysis of reverse transcription (RT)-PCR products (Fig. 5C). Correspondingly, dCas9-VP64 and MS2-P65-HSF1 expression was still detectable 20 days post-transduction (Fig. 5D). Taken together, these results indicate that SAM-induced activation of reprogramming gene expression can be performed and that endogenous expression of OCT4, LIN28, and SOX2 can be maintained for extended periods at a relatively high level, but these expression levels are not sufficient to achieve complete reprogramming of fibroblasts to iPSCs.
Discussion
In this study, we investigated the ability of CRISPR/Cas9-based ATFs to activate endogenous human pluripotency gene expression. Our results showed that the SAM system is more efficient in activating endogenous gene expression compared to the CRISPRa system. The superiority of the SAM system is in agreement with former studies optimizing CRISPR/Cas9 ATFs by combining diverse kinds of activators in one single complex (Chavez et al., 2015; Konermann et al., 2015). For example, besides the SAM system, 10 tandem copies of VP16 motifs (VP160) (Cheng et al., 2013), 12 repeated VP16 (VP192) (Balboa et al., 2015), and tripartite activator VP64-p65-Rta (VPR) (Chavez et al., 2015) have been fused with dCas9 to achieve enhanced transcription activation. In addition, a recently reported peptide, tail SunTag, fused with dCas9 can recruit multiple copies of ATFs to one single complex and demonstrate improved activation (Tanenbaum et al., 2014).
Using a mixture of three gRNAs to activate the reporter construct of OCT4 in HepG2 cells, we obtained improved activation efficiency, which is consistent with previous reports (Cheng et al., 2013). However, the two gRNA amounts (33 and 100 ng) used for each transfection had the same efficiency of activating luciferase expression, while linear relationship between gRNA amount and activation efficiency was reported previously (Cheng et al., 2013). Our observation might be due to the fact that 33 ng of gRNA plasmid is already saturating the transcriptional machinery.
The efficiency of lentivirus transduction in human fibroblasts is usually higher than 90% (Chen et al., 2003), also shown in this study (Supplementary Fig. S1). In our experiments, the activation of OCT4 expression was comparable between the SAM fibroblasts expressing the OCT4 gRNAs or KLMOS gRNAs (Fig. 3B). This suggests that transduction with a pool of gRNAs did not dilute or adversely affect the effect of using a sgRNA. Although we can detect a long-term activation of OCT4, SOX2, and LIN28A, the derivation of iPSCs using the current method reported in this study needs further optimization in regards to augmentation of the expression levels of the targeted pluripotency genes.
First, we have achieved a 60- to 100-fold transcription activation compared to fibroblasts, but this activation level is normalized to the expression level in the fibroblasts, which express pluripotency genes at very low levels. Consistent with this, we did not detect a significant increase in protein levels of these three genes (OCT4, SOX2, and LIN28A) by flow cytometry (Supplementary Fig. S2). Thus, future improvements will be focusing on further improvement of the activation efficiency. In this study, all gRNAs were designed to target the proximal promoter region. Future studies should use more gRNAs per gene, and target other regulatory regions such as enhancers.
Second, although cells were transduced by lentivirus with a MOI of 0.9, considering the pool of KLMOS gRNAs (13 gRNAs), codelivery of all gRNAs into the same cells decreases accordingly. One possibility for improvement is to generate vectors carrying all gRNA expression cassettes in one single vector as we have developed recently (Vad-Nielsen et al., 2016). However, this strategy is technically challenging for gene delivery through lentiviral vector. The presence of repetitive genomic sequences (e.g., the gRNA scaffold and U6 promoter) will lead to recombination during lentivirus generation. Thus, a combination of all-in-one multiplexed gRNA expression vector and efficient transfection methods such as nucleofection should be investigated in future studies.
Third, we recently discovered that fibroblasts that have been propagated in vitro for over 1 month showed reduced reprogramming efficiency (Zhou et al., 2016). Although we have chosen fibroblasts, which had been tested for reprogramming, the derivation of SAM fibroblasts from the parental cells might potentially affect reprograming. Thus, we recommended that codelivery of SAM and gRNAs in an all-in-one construct should be investigated in future studies for testing the generation of iPSC by SAM.
CRISPR/Cas9-based ATFs have been shown to regulate lineage commitment of cells via reprogramming of MEFs into skeletal myocytes (Chakraborty et al., 2014), stimulating neuronal differentiation from iPSC (Chavez et al., 2015), converting mouse EC cells (ESC) into extra embryonic lineages (Wei et al., 2016) and mediating human ESC differentiation into specific lineages (Kearns et al., 2014). Reprogramming of primary human fibroblasts into iPSCs has been achieved by replacing transgenic OCT4 with ATFs based on CRISPR/Cas9 (Balboa et al., 2015) or TALE (Gao et al., 2014) strategies, while exogenous MYC, KLF4, and SOX4 were still required. Therefore, complete endogenous reprogramming into iPSC by ATFs still remains a challenge. In this study, although the SAM system was able to induce continuous expression of KLMOS in fibroblasts, phenotypic change toward ES-like colony growth was not observed. The effects of activation of the endogenous pluripotency genes by these ATFs might result in partial reprogramming such as mesenchymal to epithelial transition (Koche et al., 2011), which should be investigated in the future. Taken together, existing research, including our study, indicates that complete reprogramming requires stoichiometric and temporally restricted expression of pluripotency factors.
In conclusion, we utilized CRISPR/Cas9-based ATFs to activate the expression of endogenous human pluripotency genes in various human somatic cell types. This RNA-guided gene activation can be maintained over extended periods of time. Although, the RNA-guided activation could not immediately induce reprogramming of somatic cells into iPSCs, our results provide valuable guidelines and suggestions for the generation of human iPSCs based on ATFs in the future.
Footnotes
Acknowledgments
We are grateful to Lin Lin, Yong Liu, Trine Skov Petersen, and Bettina Hansen (Department of Biomedicine, Aarhus University) for their technical advice and support. This project is supported by grants from Det Frie Forskningsråd (Danish Council for Independent Research)—DFF-1337-00128, DFF-1335-00763A (Y.L.), Lundbeckfonden (Lundbeck Foundation)—R173-2014-1105 (Y.L.), R151-2013-14439 (L.B.), European Union 7th Framework Program (PIAP-GA-2012-324451-STEMMAD), and Innovation Fund Denmark “Brainstem” (4108-00008B). K.X. and Y.Z. are financially supported by the China Scholarship Council, as well as a PhD scholarship to Y.Z. from the HEALTH faculty of Aarhus University. K.X. is further supported by the programme of excellence 2016 (Copenhagen as the next leader in precise genetic engineering CDO2016: 2016CDO04210) from the University of Copenhagen.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.