
Abstract
Abstract
The aim of the present study was to compare transgenic cells, containing human insulin gene kept under the control of mammary gland-specific buffalo beta-lactoglobulin promoter, and their counterparts, that is, nontransgenic cells, for examining their potential for the production of embryos following somatic cell nuclear transfer (SCNT). The gene construct was delivered into buffalo fetal fibroblasts (BFF) by nucleofection following which, the transfected cells were selected by culture in the presence of G418 for 3 weeks. Transgene integration into BFF genome was confirmed by polymerase chain reaction (PCR) and reverse transcriptase PCR. At passage 8–10, the growth rate, cell proliferation rate, and quantitative expression of certain genes were compared between transgenic and nontransgenic cells. The growth rate and cell proliferation rate was significantly lower (p < 0.05) for transgenic than for nontransgenic cells. Using quantitative real-time PCR it was found that the expression level of CASPASE 3, CASPASE 9, BAX, and P53 was significantly higher (p < 0.05) and that of HDAC1 and IGF-1R was significantly lower (p < 0.05) in transgenic compared with nontransgenic cells. The differences in the relative expression level of BCL-XL, MCL-1, DNMT1, DNMT3a, GDF9, FGF2, and G6PD between the two groups were not significant. Furthermore, when the two cell types were used as donor cells for production of embryos by handmade cloning, the blastocyst rate was significantly lower (p < 0.05) with transgenic (35.69% ± 1.78%) than with nontransgenic cells (48.75% ± 2.38%). In conclusion, these results indicate that differences were present between transgenic and nontransgenic cells, which may affect the efficiency of SCNT when used as donor cells.
Introduction
F
Insulin, a peptide hormone, is essential for metabolism and utilization of energy from the nutrients ingested, particularly glucose. It is pharmaceutically the most important protein used to treat diabetes, which is a major worldwide problem. Therefore, we aimed to produce buffalo transgenic cells containing human insulin (hINS) gene and use them as donor cells for generating transgenic embryos. These could eventually be used for the production of transgenic animals capable of secreting hINS in their milk. For the application of transgenic technology to large livestock animals, buffalo was the animal of choice in our study since it is the most important dairy animal in India and is capable of providing high amounts of milk. To our knowledge, this is the first study done to investigate the differences between transgenic and nontransgenic buffalo fetal fibroblasts (BFFs) and on the production of buffalo transgenic cloned embryos containing hINS gene.
The characteristic features of transgenic cells produced depend on the techniques used, such as preparation, transfection, and selection of transfected donor cells. Several passages are required for the selection procedures, which lead to higher probability of occurrence of genetic and epigenetic changes. Comparatively, the cells cultured for short time periods are more easily reprogrammable than the cells cultured for long durations (Roh et al., 2000). In addition, changes in the expression pattern of chromatin remodeling proteins in cells on account of different culture conditions could result in wide range of blastocyst and pregnancy rates (Rideout et al., 2000; Roh et al., 2000).
DNA methylation and histone acetylation play a major role in the remodeling process of the genome, and are linked with imprinting of genes in the early embryo (Grandjean et al., 2001). The DNA methyltransferase 3 (DNMT3) family establishes the de novo CpG methylation pattern and DNA methyltransferase 1 (DNMT1) maintains this pattern during chromosome replication (Chen and Li, 2006) and repair (Mortusewicz et al., 2005). Histone acetylation is responsible for chromatin relaxation, whereas, this reaction is reversed catalyzed by histone deacetylases (HDACs), which are responsible for repacking of the chromatin, and thus, leading to gene silencing (McGraw et al., 2003).
Another factor, which may affect the transgenic animal production efficiency, is the expression pattern of developmental genes in cells (Tsunoda and Kato, 2002). Insulin-like growth factor (IGF-1), also known as somatomedin C, enhances the growth-promoting activity of growth hormone. IGF-1 exerts its action through the IGF-1 receptor (IGF-1R). Basic fibroblast growth factor-2 plays an important role in cell growth, differentiation, migration, and survival in different cells and organ systems.
Also, the apoptosis of donor cells influences the cell viability, which in turn, affects the SCNT efficiency. Apoptosis is a natural defense mechanism, which promotes programmed cell death (PCD), and helps remove the damaged cells. In this context, BCL-2 family proteins are important intracellular regulators of PCD, which modulate the activation of procaspases, such as Caspase 3 and Caspase 9. Members of this family, such as BCL-2, BCL-XL, MCL-1, which are antiapoptotic, block the release of cytochrome C from mitochondria, whereas, other members, such as BAX and BAK, stimulate cytochrome C release from mitochondria, thus making them proapoptotic.
To address these issues, the present study was designed to compare transgenic and nontransgenic BFFs in terms of various parameters related to cell growth and proliferation, and expression level of several apoptosis-, epigenetic-, and development-related genes. Finally, we compared the developmental competence of embryos produced by handmade cloning (HMC) following use of transgenic and nontransgenic BFFs as donor cells.
Materials and Methods
Unless otherwise mentioned, all of the chemicals were obtained from Sigma Chemical Company (St. Louis, MO). Fetal bovine serum (FBS) used for donor cell preparation was from HyClone (Logan, UT). All media were purchased from GIBCO (Grand Island, NY) and disposable plastic wares from Nunc (Roskilde, Denmark). Ethical approval was not required as buffalo fetus was obtained from an abattoir.
DNA construction
An expression vector “pAcISUBC” was constructed having a hINS gene inserted between DNA fragments containing buffalo beta-lactoglobulin (buBLG) promoter and buBLG 3′UTR into pAcGFP-N1 (Clontech Laboratories, Inc. Mountain View, CA) vector backbone. Construction and assessment of the mammary gland-specific expression vector had been carried out earlier (Kaushik et al., 2014). Schematic representation of the recombinant plasmid pAcISUBC is shown in Supplementary Figure S1 (Supplementary Data are available online at www.liebertpub.com/cell).
Isolation, culture, and characterization of somatic cells
Primary cell culture of fetal fibroblasts obtained from Murrah buffaloes were established as described earlier (Shah et al., 2009). The cells were characterized by examining the expression of cytoskeletal proteins, vimentin, tubulin, and cytokeratin-18, by immunofluorescence staining as described earlier (Selokar et al., 2012). The cells were further characterized by reverse transcription–polymerase chain reaction (RT-PCR) and PCR for which RNA was isolated from BFFs using TRIzol (Invitrogen, Carlsbad, CA) reagent as per the manufacturer's protocol.
After treatment with DNase, a RT reaction was performed for the synthesis of cDNA using superscript reverse transcriptase III (Invitrogen). The expression of cell-specific markers (VIMENTIN, TUBULIN, FSP, DESMIN, CYTOKERATIN-8, and CYTOKERATIN-18) was analyzed by RT-PCR. The RT-PCR was performed using the following program: initial denaturation at 95°C for 3 min, followed by 95°C for 30 s, annealing temperature for 30 s, 72°C for 30 s for 39 cycles, and 72°C for 10 min in the last cycle. For sex determination, genomic DNA was isolated and PCR was performed for the genes PLP and SRY with the following conditions: 94°C for 30 s (denaturation), 56°C for 30 s (annealing), and 72°C for 30 s (extension). The primer sequences used for cell characterization are given in Supplementary Table S1.
Nucleofection and Selection
BFFs were thawed and grown to 70%–80% confluence in DMEM supplemented with 10% FBS, after which the cells were separated by trypsinization. Before transfection, pAcISUBC plasmid was linearized with Afl(II) enzyme. For nucleofection, 6 μg of plasmid DNA was added to 0.5–1 × 106 cells that had been resuspended in P3 solution and supplement 1 (AMAXA Biosystems, Gaithersburg, MD). The cell/DNA mixture was nucleoporated in 1-cm transfection cuvettes using EN-150 program. The cells were incubated in prewarmed culture media for 10 min at 37°C and the cell suspension was transferred to 35 × 10-mm cell culture Petri dishes containing DMEM supplemented with 10% FBS. After 24 h of nucleofection, expression of enhanced green fluorescent protein (EGFP) was detected using an inverted fluorescence microscope (Nikon, Tokyo, Japan).
For determining the transfection efficiency, the cells were fixed 24 h after transfection in 4% paraformaldehyde for 30 min and stained with Hoechst 33342 (10 μg/mL) for 30 min; then, 10 different fields (200 × ), which were chosen randomly, were examined using an epifluorescence microscope, and the number of transfected (GFP positive) and total (Hoechst stained) cells was determined. For the selection, the cells were exposed to 800 μg/mL geneticin (G418) for 7 days, followed by exposure to 400 μg/mL G418 for 14 days to obtain transfected cell colonies. The transfected cell colonies were picked up with the help of cloning rings (Sigma; St. Louis, MO) and were cultured in a 24-well plate. The transfected cells were expanded further by subpassaging up to 10 times (Fig. 1).

Nontransfected cells under bright light
Detection of transgene integration
For detection of transgene integration in BFFs, genomic DNA was extracted and PCR amplification was carried out using primer set (hINS forward and hINS reverse) that amplifies a 275 bp fragment. PCR conditions consisted of initial denaturation at 95°C for 5 min, followed by 30 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 30 s, and final extension at 72°C for 10 min. After amplification, the amplified PCR product was analyzed on 2% agarose gel. The oligonucleotide sequence used is shown in Supplementary Table S2.
Analysis of GFP expression
For further confirmation of the presence of the gene construct in transgenic cells, RNA was isolated from transgenic and nontransgenic cells using TRIzol reagent and cDNA was prepared. RT-PCR was performed using the primer set, GFP forward and GFP reverse. PCR conditions consisted of initial denaturation at 95°C for 5 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and final extension at 72°C for 10 min. The products obtained were run on 2% agarose gel. The primer sequence used is shown in Supplementary Table S2.
Examination of relative cell viability and proliferation rate of transgenic and nontransgenic cells
The relative cell viability of transgenic and nontransgenic cells was determined by MTT [3-(4, 5-di-methylthiazol-2-yl) 2, 5- diphenyl tetrazolium bromide] assay as described previously (Selokar et al., 2012). In brief, 2 × 103 cells between 8 and 10 passage number were seeded in 96-well plates and cultured in DMEM supplemented with 10% FBS for 72 h.
The cells were then incubated with 5 mg/mL MTT at 37°C for 2 h following which dimethyl sulfoxide (DMSO), diluted with culture medium (1:1), was added and the reactants mixed thoroughly until the formazan crystals dissolved completely. The optical density of the dissolved formazan was measured at 570 nm through NanoQuant spectrophotometer. The absorbance ratio of transgenic to nontransgenic cells was calculated and presented as relative cell viability of transgenic cells. For calculating the proliferation rate, 3 × 104 cells were seeded in each 60-mm Petri dish. The cells were cultured for 120 h and harvested after every 24 h for determination of total cell number. Live and dead cell count was calculated by the dye exclusion method, using Trypan Blue dye and a Neubauer's hemocytometer. The percentage of live cells is given in Supplementary Table S3. The experiment was repeated three times.
Gene expression analysis of cells
Isolation of RNA from transgenic and nontransgenic cells was carried out using TRIzol reagent according to the manufacturer's instruction. Following DNase treatment, a RT reaction was performed for the synthesis of cDNA using superscript reverse transcriptase III (Invitrogen). Quantitative real-time PCR (qPCR) was done according to the procedure described earlier (Singh et al., 2014). mRNA was quantified through qPCR using CFX96 C1000 Thermal Cycler (Bio-Rad, Hercules, CA). The reaction mixture (10 μL) contained 5 μL of SYBR Green Master Mix (Maxima SYBR Green Mastermix, Thermo Scientific), 0.2 μL of 10 μM of each primer, and cDNA obtained from both cell types. Thermal cycling conditions for the genes consisted of initial denaturation for 3 min at 95°C, followed by 40 cycles of 15 s at 95°C, corresponding annealing temperature for 15 and 15 s at 72°C, followed by final extension of 15 s at 72°C.
The list of primer sequences used is given in Supplementary Table S4. All the primers were confirmed for their PCR efficiencies, whereas the specific products were checked through melt curve analysis, and for the detection of size, the products were run on 2% agarose gel. Relative gene expression was determined using 2−ΔΔCt method, where DCt = Ct (target gene)—Ct (internal reference) and DDCt = DCt sample—DCt calibrator. Here, nontransgenic cells were taken as calibrator for each gene and glyceraldehyde 3-phosphate dehydrogenase served as the internal reference gene. The experiment was repeated thrice, each time using three replicates of both the cell types.
HMC and assessment of embryo development
Transgenic and nontransgenic donor cells were prepared by growing them in DMEM to full confluence so to have them in G1 stage of the cell cycle, as previously described (Selokar et al., 2012). The recipient demicytoplasts were prepared through a series of procedures, which included in vitro maturation, cumulus/zona pellucida removal, and enucleation of oocytes using microblade as described earlier (Shah et al., 2009). Furthermore, fusion, activation of reconstructs, and in vitro culture (IVC) of nuclear-transferred (NT) embryos were carried out as described previously (Shah et al., 2009). The blastocyst rate, taken as a measure of the developmental competence of NT embryos, was recorded on day 8 of IVC.
Statistical analysis
The relative cell viability, proliferation rate, and expression level of different genes in the transgenic and nontransgenic cells, and the developmental competence of embryos produced from these cells were compared by Student's t-test followed by post hoc test (least significant different), using a statistical software program (SPSS 20, 2011, IBM). The results were presented as mean ± standard error of the mean. The differences were considered to be significant at p < 0.05.
Results
Establishment and characterization of BFFs
BFFs were successfully isolated and established in culture. When the cells were characterized by examining the expression of cell-specific markers by immunofluorescence staining, they were found to be vimentin and tubulin positive and cytokeratin-18 negative (Supplementary Fig. S2A). The cells were also characterized through RT-PCR and were found to be positive for VIMENTIN, TUBULIN, FSP, and DESMIN, and negative for CYTOKERATIN-8 and CYTOKERATIN-18 as shown in Supplementary Figure S2B. When the cells were subjected to PCR for determination of sex, the expression of X chromosome-specific gene PLP, but not that of Y chromosome-specific gene SRY was detected, whereas the control male fibroblasts showed expression for both PLP and SRY genes (Supplementary Fig. S2C).
Transfection of pAcISUBC plasmid vector into BFFs and selection of transfected cells
BFFs were transfected with the pAcISUBC plasmid containing hINS gene kept under the control of mammary gland-specific buBLG promoter. The efficiency of transfection as observed under fluorescence microscope was found to be 73.6% ± 1.36%. The transfected cells were selected by culture in the presence of G418 for 2 weeks, following which the G418-resistant colonies were picked up with cloning rings and expanded further. The transfected cells thus obtained were cryopreserved for further use.
Transgene integration detection
The presence of gene construct containing hINS gene in the transfected cells was detected through PCR amplification. The PCR products run on 2% agarose gel showed a band at 275 bp in the lane in which DNA from transfected cells were loaded, whereas no band was observed in case of nontransfected cells (Fig. 2A). Also, RT-PCR showed the presence of GFP expression in transfected cells, further confirming the presence of the gene construct in the transfected cells (Fig. 2B).

Identification of transgene integration by PCR analysis
Comparison of relative cell viability and proliferation rate of transgenic and nontransgenic cells
The proliferation rate of transgenic cells was found to be significantly lower (p < 0.05) than that of nontransgenic cells (Fig. 3A). Also, the relative cell viability determined by MTT assay was significantly lower (p < 0.05) for transgenic compared with nontransgenic cells (Fig. 3B).
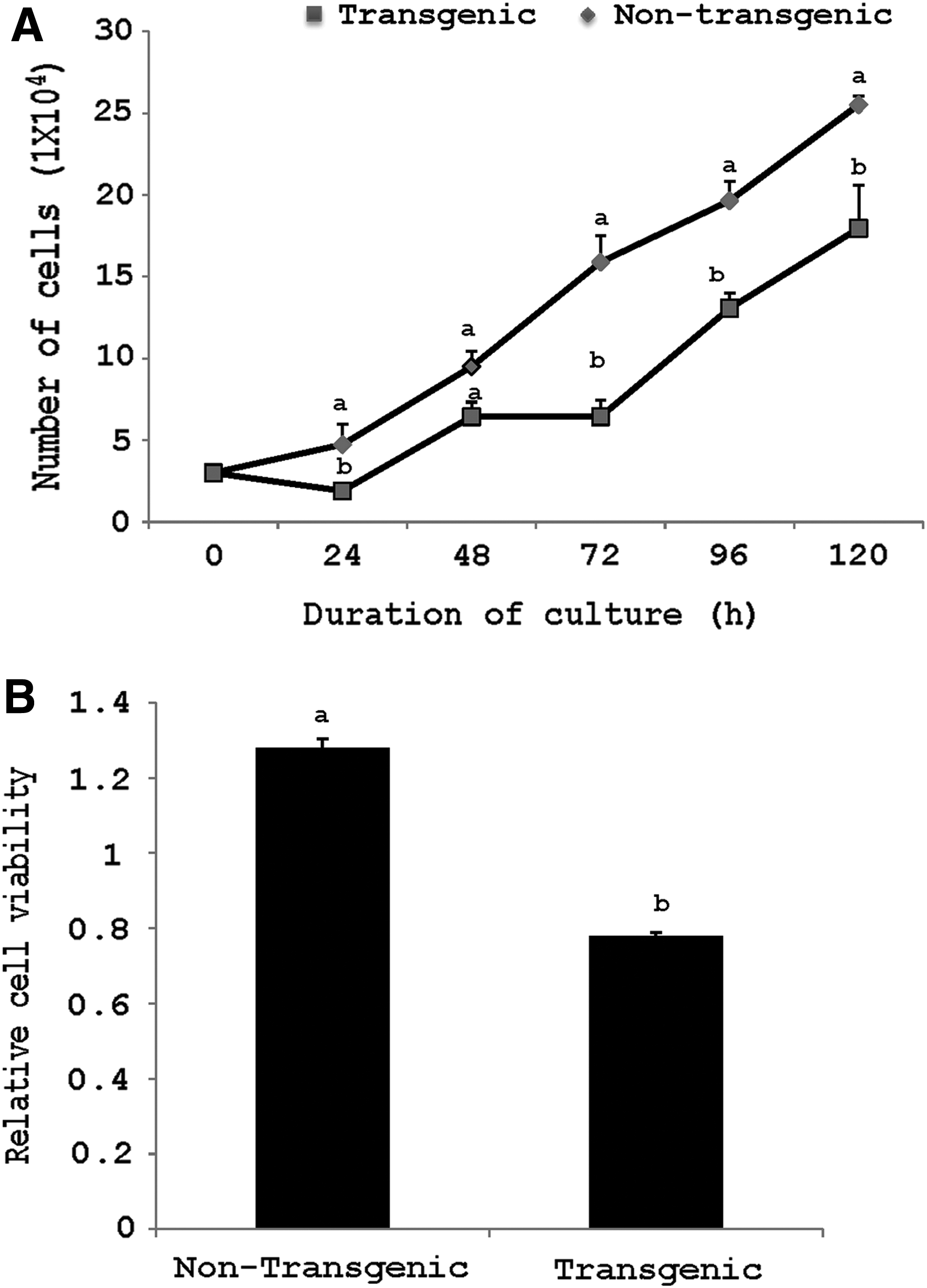
Comparison of relative cell proliferation of transgenic and nontransgenic fetal fibroblast cells for 120 h in a 24-well dish
Gene expression analysis in transgenic and nontransgenic cells
Comparison of the relative gene expression in the transgenic and nontransgenic cells revealed that among the apoptosis-related genes, the relative expression levels of CASPASE 3, CASPASE 9, BAX, and P53 was significantly higher (p < 0.05) in transgenic than in nontransgenic cells. However, there was no significant difference in the expression level of BCL-XL and MCL-1 between the two groups (Fig. 4A). Among epigenetic-related genes, the expression level of HDAC1 was significantly higher (p < 0.05) in nontransgenic than in transgenic cells (Fig. 4B), but that of DNMT1 and DNMT3a was similar between the two groups. Among the development-related genes, the expression level of IGF-1R was significantly higher (p < 0.05) in nontransgenic compared with transgenic cells, whereas that of GDF9, FGF2, and G6PD was not significantly different between the two cell types (Fig. 4C).
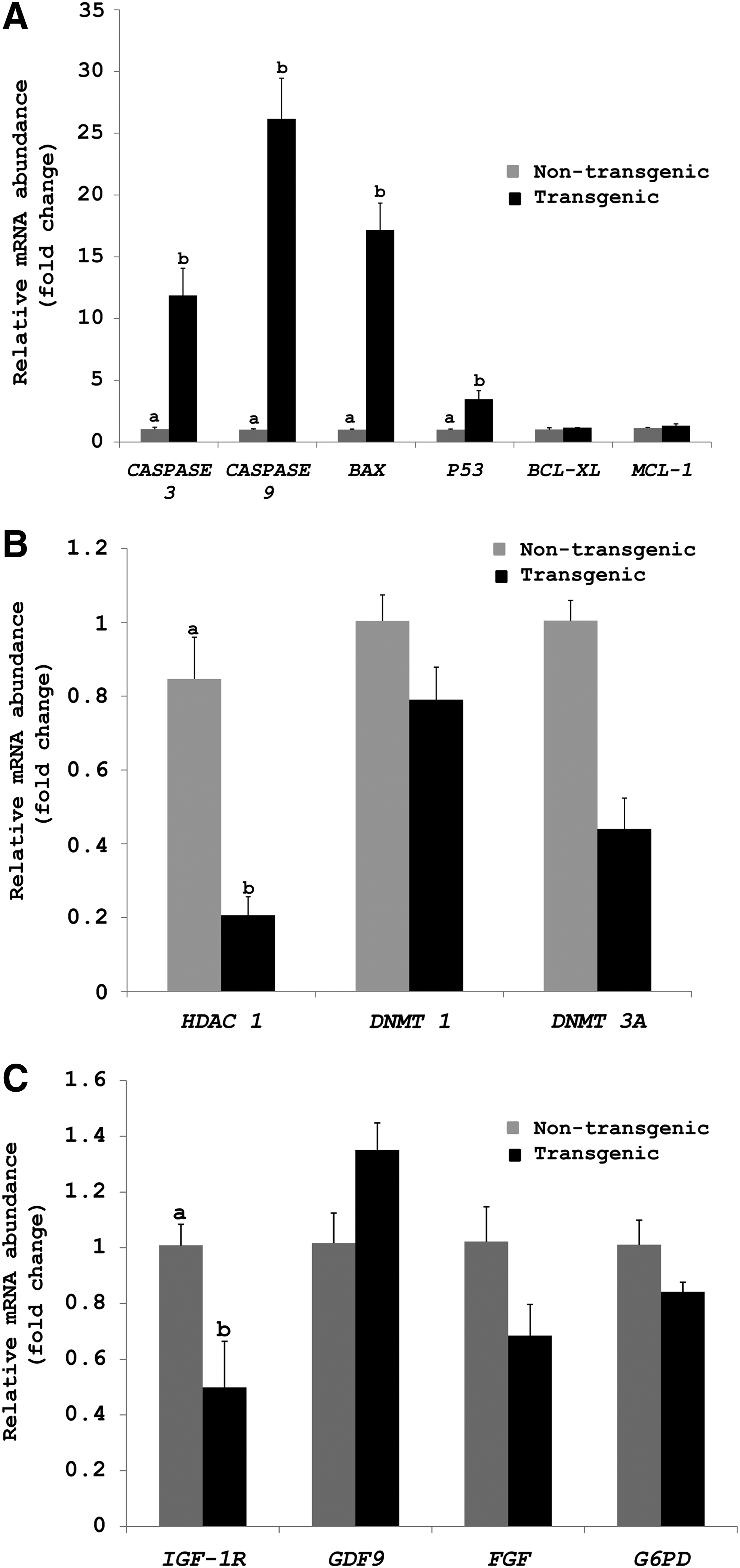
Relative expression level of apoptosis- related
Developmental competence of transgenic and nontransgenic embryos
Embryos were successfully produced by HMC using transgenic cells containing hINS gene as donor cells, and EGFP fluorescence was successfully detected in these embryos (Fig. 5). The rate of development of cloned embryos produced from transgenic and nontransgenic cells is presented in Table 1. The blastocyst rate was significantly higher (p < 0.05) for the embryos produced from nontransgenic cells than those derived from transgenic cells (48.75% ± 2.38% vs. 35.69% ± 1.78%) although the cleavage rate was not significantly different.

GFP expression in buffalo transgenic embryos under bright field
Blastocyst rate data are from 15 trials. Values are mean ± SEM. Values with different superscript within the same column differ significantly (p < 0.05).
HMC, handmade cloning; SEM, standard error of the mean.
Discussion
Genetically modified animals play a remarkable role in agriculture and biomedical area, particularly in the production of valuable pharmaceutical proteins and the supply of xenografts. In our study, fetal fibroblasts were chosen because they are rapidly growing cells and have potential for numerous cell divisions before becoming senescent in culture (Baguisi et al., 1999; Cibelli et al., 1998; Schnieke et al., 1997). These characteristics make them a suitable candidate for transfection experiments. We isolated somatic cells from fetus and characterized them by examining the presence of certain cell-specific markers. Immunofluorescence staining revealed that the cells expressed vimentin and tubulin, but not cytokeratin-18, suggesting that they were of fibroblast origin. Their fibroblast origin was further confirmed by the presence of VIMENTIN, TUBULIN, FSP, and DESMIN, but not CYTOKERATIN-8 and CYTOKERATIN-18 by RT-PCR.
Earlier studies suggest that nucleofection offers higher transfection efficiency as compared with other transfection methods when used in primary cell cultures (Dityateva et al., 2003; Ganesh et al., 2003), mouse and human embryonic stem cells (Hohenstein et al., 2008), and also cell lines which are difficult to transfect (Martinet et al., 2003). Reports are also available on successful production of cloned transgenic porcine blastocysts using cells transfected through nucleofection (Nakayama et al., 2007; Skrzyszowska et al., 2008). Another report demonstrated that with the exception of HEK 293 cells, nucleofection showed less toxicity and more efficiency at delivering DNA into adherent cells such as primary fibroblasts and embryonic stem cells, and cells in suspension such as lymphoblasts and primary hematopoietic stem cells (Maurisse et al., 2010). Therefore, we chose nucleofection for the production of transgenic cells containing hINS gene.
Progress made in animal cloning through SCNT has made the production of transgenic animals possible using genetically altered cells. We compared several characteristics of transgenic cells with nontransgenic cells assuming that the genetic material stably integrated into the fetal fibroblasts and their selection using G418 could alter the cell growth and viability, and the level of apoptosis, which in turn, could lead to lower production rate of transgenic compared with nontransgenic embryos. The proapoptotic genes, CASPASE 3 and CASPASE 9, are responsible for the release of cytochrome C from mitochondria thus leading to intrinsic cell death (Brentnall et al., 2013).
In our study, it was observed that the transgenic cells showed significantly higher (p < 0.05) expression level of these proapoptotic genes compared with nontransgenic cells. P53 is a tumor-suppressor protein, which mediates the checkpoints induced through DNA damage through the transactivation of a number of growth inhibitors and apoptosis genes (Ko and Prives, 1996). The transgenic cells had significantly higher (p < 0.05) expression level of P53 compared with nontransgenic cells, which could be because of activation of these genes due to higher level of transfection-induced DNA damage.
The Bcl-2 family comprises of members having dual functions in apoptosis. For instance, BCL-2, BCL-XL, and MCL-1 are antiapoptotic, whereas BAX and BID are proapoptotic (Zinkel et al., 2006). The transcript level of BAX, another proapoptotic gene was also found to be significantly higher (p < 0.05) in transgenic than in nontransgenic cells. However, the expression level of two antiapoptotic genes examined in our study, that is, BCL-XL and MCL-1, was similar between transgenic and nontransgenic cells. These observations suggest that the transfection of donor cells might have weakened their ability to avoid apoptosis and activated pathways leading to PCD.
These observations are further supported by the results of relative cell viability measured by MTT assay and the cell proliferation rate, both of which were found to be significantly lower (p < 0.05) for transgenic compared with nontransgenic cells. Our results are in agreement with those of Li et al. (2012), who reported that the transfected cells tend to undergo CASPASE 3 activation which induces cell death. Similarly, another study reported that tetracycline on/off-transfected cells showed a lower relative expression of antiapoptotic BCL-2, whereas triple transfected cells showed a higher expression level of P53 and BAX in comparison to their respective nontransfected counterparts (Kim and Hyun, 2014).
Variations exist in cloning efficiency between transgenic and nontransgenic cells following their use as donor cells due to altered gene expression pattern in transfected cells. In fact, variations observed in the expression profile of epigenetic-related genes in embryos and cloned animals are suggested to be linked with the reprogramming process (Beyhan et al., 2007; Dean et al., 2001; Suzuki et al., 2008). DNA methylation is one of the most important epigenetic events. Methylation of DNA is established and maintained by DNA methyltransferases (DNMTs). DNMT1 is a maintenance methyltransferase, which adds a methyl group to the newly formed DNA strand during S phase of cell cycle, whereas, DNMT3a is responsible for the establishment of de novo methylation during gametogenesis and early embryonic development (Bestor, 2000; Okano et al., 1999). It has been reported that using donor cells with lower expression level of maintenance DNMTs, may enable the process of reprogramming in the reconstructed embryos (Beyhan et al., 2007).
In our study, no significant difference (p < 0.05) was observed in the transcript level of DNMT1 and DNMT3a in transgenic and nontransgenic cells. Another important factor responsible for epigenetic regulation is histone acetyltransferases, which adds acetyl group to histone proteins, thus neutralizing its negative charge, which in turn weakens its bond with negatively charged DNA. This process is reversibly catalyzed by HDACs. Therefore, hypoacetylated histones are related to lower transcriptional activity due to closed chromatin structure formation (Rice and Allis, 2001). In our study, the expression level of HDAC1, which was found to be lower in transgenic than in nontransgenic cells, could not be correlated with reduced developmental potential of transgenic embryos. Our results are contradictory to those of Beyhan et al. (2007), who reported that the expression levels of HDAC1 in donor cells are related to the potential of embryos to develop to different embryonic stages.
The present study demonstrates that the developmental potential of cloned embryos produced from transgenic cells is significantly lower (p < 0.05) than that of embryos produced from nontransgenic cells despite their similar origin. These results are in agreement with those of several earlier reports which stated that there were significant differences in the efficacy of generating embryos when transgenic and nontransgenic cells were used as nuclear donors (Arat et al., 2002; Bhuiyan et al., 2004). Huang et al. (2010) reported that no NT embryos were produced when EGFP-positive BFFs were taken as nuclear donor.
However, developmental rate of transgenic and nontransgenic embryos were found to be similar in other studies (Brink et al., 2000; Cibelli et al., 1998; Roh et al., 2000). It was demonstrated in another report that in spite of genetic manipulation of bovine oviductal epithelial cells, the developmental competence was not compromised (Gong et al., 2004). Similar observations were made when transgenic and nontransgenic goat fetal fibroblasts were used (Iguma et al., 2005; Zhang et al., 2010). The differences in results of various studies could be due to variations in species, strains, or cell types used in the respective studies.
In conclusion, this study demonstrates that transgenic cells exhibit reduced relative cell viability and proliferation rate and altered gene expression pattern compared with nontransgenic cells. These characteristics of transgenic cells may be important determinants of the reduced developmental potential of NT embryos produced from these cells.
Footnotes
Acknowledgments
This work was supported by grant BT/PR8725/AAQ/1/556/2013 from the Department of Biotechnology, Government of India, New Delhi, India. Parul Mehta and Ankur Sharma were supported by CSIR and ICMR fellowships, respectively.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.