
Abstract
Abstract
We designed a study to induce differentiation of Oct4-GFP (expression of Green Fluorescent Protein of oct4) embryonic stem cells (ESCs) by embryoid body (EB) culture system into germ cells (GCs) using retinoic acid (RA) and evaluated the expression level of (Fkbp6, Mov10l1, 4930432K21Rik, and Tex13) in differentiated cells. The expression levels of four GC-related genes, Oct4, Mvh, Scp3, and Stra8, was determined by quantitative real-time polymerase chain reaction (q-RT-PCR). Immunostaining and flow cytometry were used as additional tests to confirm q-RT-PCR findings. A significant increase occurred in the expression of meiotic markers and specific genes, Fkbp6 (p = 0.00), Mov10l1 (p = 0.01), and Tex13 (p = 0.00) in ESCs treated with RA (+RA) compared with the controls (−RA). Oct4 expression was decreased in all studied groups. The expression levels of 4930432K21Rik, Mvh, Stra8, and Scp3 in the +RA group was higher than that of the −RA group. Flow cytometry analysis showed that mean number of Mvh-positive cells in the +RA group was greater as compared with ESCs, −RA and EB7 groups (p = 0.00). Downregulation of Oct4 as a pluripotency factor as well as the expression of meiosis markers, this hypothesis is raised that ESCs are differentiated by RA, and have been introduced into the zygote/pachytene of first meiosis as GC-like cells.
Introduction
D
Whether a meiosis-inducing factor (Byskov et al., 1995) requires PGCs to enter meiosis or if it is an autonomous event (Mclaren, 1997) is still a controversial issue. Over the past several decades, researchers have attained significant results in designing an appropriate in vitro model for the differentiation of embryonic stem cells (ESCs) into GCs (Hayashi et al., 2011). It seems that these ESC-derived PGCs have the ability to enter meiosis as male and female gametes. However, compared with in vivo, they do not undergo normal meiosis or become a functional gamete (Sun et al., 2014).
Defects in natural and complete meiosis are one of the obstacles in achieving functional gametes. ESCs derived from the blastocyst inner cell mass (ICM) in vitro are pluripotent stem cells that can accumulate and form cell clusters or embryoid bodies (EBs). Under certain conditions these cells can differentiate into all three embryonic germ layers (Li et al., 2016).
Results from genital ridge studies and epiblast-derived PGCs in vitro indicate that these cells respond to signals, such as leukemia inhibitory factor (LIF), stem cell factor (Matsui et al., 1991), tumor necrosis factor-α, forskolin, basic fibroblast growth factor (bFGF), macrophage growth factor, and kit ligand to promote proliferation (Kawase et al., 1994). In addition, retinoic acid (RA)-induced intracellular signals also play an important role in GC differentiation (Teletin et al., 2017). The proliferative function of RA may be due to the induction of antiapoptotic signals or the shortening of the PGC cycle (Pesce et al., 1993). RA induces Stra8 (stimulated by RA 8) expression in PGCs. Stra8, which is continuously expressed at the premeiosis GC stage, is one of the essential factors for the onset of meiosis in both males and females (Kocer et al., 2009).
Recently, Endo et al. showed that the RA-STRA8 signal not only regulates the onset of meiosis but also is effective in differentiating spermatogonia (Endo et al., 2015). RA differentiated (Aa1) spermatogonia into (A1) spermatogonia in the embryonic testis (Snyder et al., 2010). Additionally, researchers have investigated direct differentiation of somatic cells, such as bone marrow stem cells, into GCs (Kashani et al., 2014). Other studies have shown the production of male GCs from mouse bone marrow-derived mesenchymal stem cells and embryonic carcinoma cells (ECs) under the induced effects of RA (Nayernia et al., 2006). However, RA has been known as an inducer and effector in male reproduction for many years (Wolbach and Howe, 1925).
Studies in reproductive biology of mammalian embryonic development have two major problems: limited access to cellular material and the lack of specific markers that are expressed only in germline or GCs in in vitro differentiation. Also, genetic and morphologic similarities between ESCs and PGCs make it difficult to distinguish them in culture conditions and in vitro differentiation (Toyooka et al., 2000). In recent years, many efforts have been made to determine effective regulators for the generation of PGCs and their genetic contents through in vitro cell culture (Leitch et al., 2013).
Identifying the genetic network function during the specification of GCs has also been considered by researchers (Magnusdottir et al., 2013). Fkbp6, Mov10l1, 4930432K21Rik, and Tex13 are specific markers expressed in GCs, but have low or no expression in ESCs (Sabour et al., 2011. (Mov10l1, identified in mouse spermatogonia cells, is an autosomal and GC-specific gene (Poongothai et al., 2009). Tex13 is an X-linked gene that is expressed in the process of GC specification at the onset of spermatogonia differentiation (Kwon et al., 2014). The Fkbp6 gene was obtained from RNA in mouse testicular cells by real-time polymerase chain reaction (RT-PCR) (Leitch et al., 2013).
Mutations in this gene are related to male infertility (Zhang et al., 2007). The 4930432K21Rik is a novel gene with unknown function that is expressed in PGCs, the testis, spermatozoa, and oogonia. Currently, there are few studies regarding the expression level of these genes using inducers in differentiation in vitro. To optimize and improve in vitro differentiation of GC development from ESCs, we designed a study to induce ESCs differentiation into GCs using RA, and evaluated the expression level of Fkbp6, Mov10l1, 4930432K21Rik, and Tex13 in differentiated cells.
Materials and Methods
The maintenance and care of experimental animals complies with National Institutes of Health guidelines for the humane use of laboratory animals (MUBABOL.REC.1393.7).
Animals
NMRI mice were housed and bred in the central animal house of the Babol University of Medical Science under a 12-hour light (6:00 AM to 6:00 PM) and 12-hour dark regime at 22–24°C. Mice were given food and water ad libitum.
Study design
There are three groups in this experimental study. One, ESCs differentiated using EB method for 7 days then EB aggregation singled and cultured with 2 μM RA for 7 days as (+RA); two, EB aggregation in day 7 singled and cultured without RA for 7 days as control group (−RA); three, EB aggregation in day 7 of culture as (EB7).
Culture medium
Mouse embryonic fibroblast (MEF) medium: This is a knockout Dulbecco's modified Eagle's medium (DMEM; Gibco, Paisley, UK) containing 15% FBS (Biowest), Pen/Str/Glu 100 U/mL (Gibco), 100 mM nonessential amino acids (Gibco), 0.1 mM 2-Mercaptoethanol (Gibco). ESC culture medium: KnockOut DMEM (Gibco) containing 15% KnockOut SR (Gibco), Pen/Str/Glu 100 U/mL (Gibco), 100 mM nonessential amino acids (Gibco), 0.1 mM 2-Mercaptoethanol (Gibco), and 1000 IU/mL LIF (Chemicon). EB medium: This is ESC medium without LIF. ESC differentiation medium: ESC culture medium that is supplemented with 2 μM RA (Sigma, Germany) and 15 ng/mL bFGF (Invitrogen).
Culture of MEFs
ESCs require a feeder layer for growth. For this, E13.5 mice were used. Briefly, female and male were housed together in a cage (two females with one male mouse) for mating. The morning after mating, the vaginal plugs were checked, and pregnant mice were identified and scored as E0.5. After 13 days, pregnant mice were sacrificed and the embryos were harvested. Embryos were isolated and washed. The heart and liver were separated from the embryo, crushed using an 18-gauge needle, and then cultured in MEF media. After two passages, the MEF cells were ready for inactivation by mitomycin C for subsequent use as a feeder layer for ESC culture. We used passages 2–4 in our work.
Culture and passage of mouse ESCs
The OG2 (ΔPE-GFP) ESC line (a kind gift from Dr. Sabour, Max Planck, Germany) was used in this study. OCT4 is a well-accepted pluripotency factor, playing a dominant role in pluripotency maintenance of ESCs (Masui et al., 2007). OCT4 begin to show high expression levels in the ICM, but its levels decreased as the cells entered the epiblast stage. It was cultured briefly as described below. The mouse ESC (mESC) line was maintained on mitomycin C-treated MEFs in 0.1% gelatin-coated 25-cm2 flasks in ESC medium.
For gelatinization first, gelatin 0.1% with deionized water was autoclaved. After cooling slightly the solution was poured into the culture dishes. After 40 minutes fluid was removed and culture dishes placed under the UV for 15–20 minutes. Undifferentiated ESCs were cultured at 37°C, 5% CO2, and 95% humidity, and the medium was renewed daily. Seventy-two hours after primary culture, when the colony size increased, cultures were trypsinized and expanded at a ratio of 1:3 on fresh feeder cells. The medium was changed every day.
ESC induction for EB and GC formation in vitro
To induce GC differentiation from ESCs, the protocol described by Geijsen was followed with a slight modification (Geijsen et al., 2004). Briefly, ESCs were dissociated with 0.25% trypsin-EDTA (Invitrogen) and collected in EB medium. To separate MEFs from ESCs, the ability of MEFs to reattach faster than ESCs to a tissue culture plate was used for MEF removal. After two rounds of reseeding (about 30–40 minutes), EB formation was induced in suspension culture, where ESCs were seeded at a density of 1.5 × 106 cells per cm2 in an EB differentiation media in a nonattachment 10-cm2bacterial dish. After 7 days, EBs were dissociated and digested by collagenase IV treatment (0.01%) to obtain cell suspensions. Then, being filtered and seeded in a gelatinized dish at about 20,000 cells per cm2, cells were fed daily with ESC medium supplemented with 2 μM RA and bFGF (15 ng/mL) for 7 days.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from ESCs, RA-treated cells (+RA), non-RA-treated cells (−RA), and 7-day EB (EB7), and somatic tissues of the testis and brain were isolated using the RNA Isolation Kit (Roche). Genomic DNA contamination was eliminated using DNase I. RNA quality was determined using NanoDrop 2000c (Thermo Scientific). cDNA was prepared in a total volume of 10 μL using a cDNA Synthesis Kit (TaKaRa) according to the manufacturer's protocol.
Target gene expressions were normalized based on the mouse housekeeping gene Hprt. Gene transcripts were assessed using Syber Green I PCR Master Mix (Applied Biosystems) containing 150 nmol each of the forward (F) and reverse (R) primers (Table 1). Quantitative PCR analysis was performed using the ABI 7300. Relative quantification of gene expression was calculated using the 2−ΔΔCt method. Three technical replicates were used for each quantitative RT-PCR (q-RT-PCR); a no-template blank served as a negative control. In all reactions, mouse testis and brain were used as positive and negative controls, respectively.
Immunofluorescent staining
Mvh-positive cells appear and can develop into PGC-like cells. Evaluation of these cells was performed after treatment in each group. Briefly, cells that were cultured in two-well chamber slides were fixed in 100% methanol (chilled at −20°C) at room temperature for 5 minutes. Then, cells were heated in antigen retrieval buffer (100 mM Tris, 5% [w/v] urea, pH 9.5) to 95°C in order for optimal performance of certain antibodies. Coverslips were heated at 95°C for 10 minutes. The cells were incubated for 10 minutes in phosphate-buffered saline (PBS) containing 0.1%–0.25% Triton X-100 (ICN) for permeabilization. Subsequently, cells were incubated with 1% bovine serum albumin (BSA), and 22.52 mg/mL glycine in PBST (PBS +0.1% Tween 20) for 30 minutes to block nonspecific binding of the antibodies.
Cells were then incubated in diluted Mvh primary antibody (1:100; Abcam 13840) in 1% BSA in PBST in a humidified chamber overnight at 4°C. Then, cells were incubated with the secondary antibody (goat anti-rabbit IgG-PE: sc-3739, 1:100) in 1% BSA for 1 hour at room temperature in the dark, followed by incubation with 0.1–1 μg/mL DAPI (Sigma) (DNA stain) for 1 minute. Coverslips were mounted with a drop of mounting medium. Finally, cells were evaluated under an inverted fluorescence microscope (Canada Smart).
Immunohistological examination
Slides were allowed to reach room temperature. Slides were washed three times in TPBS (PBS-Tween), each time for 5 minutes before being immersed in Triton X-100 (0.2% for cytoplasmic antigen) for 20 minutes. Blocking was performed in 10% normal serum with 1% BSA in TPBS for 2 hour at room temperature, followed by incubation with Mvh primary antibody (Abcam 13840) (1:100) diluted in TPBS with 1% BSA overnight at 4°C in the dark. Fluorochrome-labeled secondary antibody (goat anti-rabbit IgG-PE) diluted in TBS with 1% BSA was applied to the slide and the slide was incubated for 1 hour at room temperature in the dark. The coverslip was mounted using a compatible mounting medium.
Flow cytometry
Cells were fixed before intracellular staining to ensure stability of soluble antigens or antigens with a short half-life. Fixation occurred by placing the cells in 0.01% formaldehyde for 10–15 minutes. One hundred microliters of detergent-based permeabilizing agent, Triton X-100 (0.1%–1% in PBS), was added and the cells were incubated in the dark at room temperature for 15 minutes. Around 0.1–10 μg/mL Mvh primary antibody (Abcam 13840) was added and the cells were incubated for at least 30 minutes at 4°C in the dark. The fluorochrome-labeled secondary antibody (goat anti-rabbit IgG-PE) was diluted in 3% BSA/PBS at the optimal dilution (1:100–1:400) and added to the cells, which was followed by incubation for at least 20–30 minutes at 4°C in the dark.
Cells were resuspended in ice cold PBS, 3% BSA, and 1% sodium azide. Secondary antibody IgG-PE was detected using the FL1 channel of the FACSCalibur™ flow cytometer (BD Biosciences) and the percentage of positive cells was measured by the FlowJo software.
Statistics
All experiments were independently repeated at least three times. Data are presented as mean ± standard deviation. Statistical analysis was determined using ANOVA. All statistical tests were performed using SPSS (Statistical Package for the Social Sciences, version 22.0; SPSS, Inc., Chicago, IL) software. Flow cytometry data were analyzed using FlowJo 7.6 software. p-values <0.05 were regarded as significant.
Results
Morphological evaluation
ESC colonies (Fig. 1A–C) were trypsinized, isolated, and seeded for the suspension culture at ∼1 × 106 to 1.5 × 106 cells/cm2 in a 10-cm nonadherent bacterial plate to form EBs for 7 days (Fig. 1D–F). EB7 were isolated and cultured in gelatinized tissue culture plate for an additional 7 days. Cell proliferation and differentiation occurred in the presence of bFGF and RA. Morphological evaluation of these cells over the duration of culturing showed that those small and single EB cells changed to round-shaped cells. Single cells in tissue culture plates attached and formed an integrated network that was tightly adhered to the bottom of the dish (Fig. 1G–I).

Morphological assessment of Oct4-GFP ESC colonies, day 7 of EB culture (EB 7), and ESC-derived GCLCs.
Additionally, we cultured EB7 cells without dissociation (aggregated EBs) in tissue culture plates for evaluation and comparison. After RA treatment for 7 days we saw that cell–cell adhesions were loose in the colonies and the cell shape changed to round-shaped cells as well as single EB cells.
RA is sufficient to induce GC-like cells
To determine the effect of 2 μM RA in regulating GC development in vitro, we cultured the mouse Oct4-GFP ESC line in the EB culture system for 7 days and continued single cell culture of EBs for an additional 7 days of differentiation by RA. The expression levels of four GC-related genes (Fkbp6, Mov10l1, 4930432K21Rik, and Tex13), and Oct4, mvh (vasa, ddx4), sycp3 (synaptonemal complex protein 3), stra8, and hprt (mouse housekeeping gene) was determined by q-RT-PCR. These findings were confirmed by determining their expression in somatic mouse tissues of the brain (as a negative control) and testis (as a positive control). Positive immunostaining and protein expression of Mvh by flow cytometry were used as additional controls to ensure proper in vitro GC differentiation.
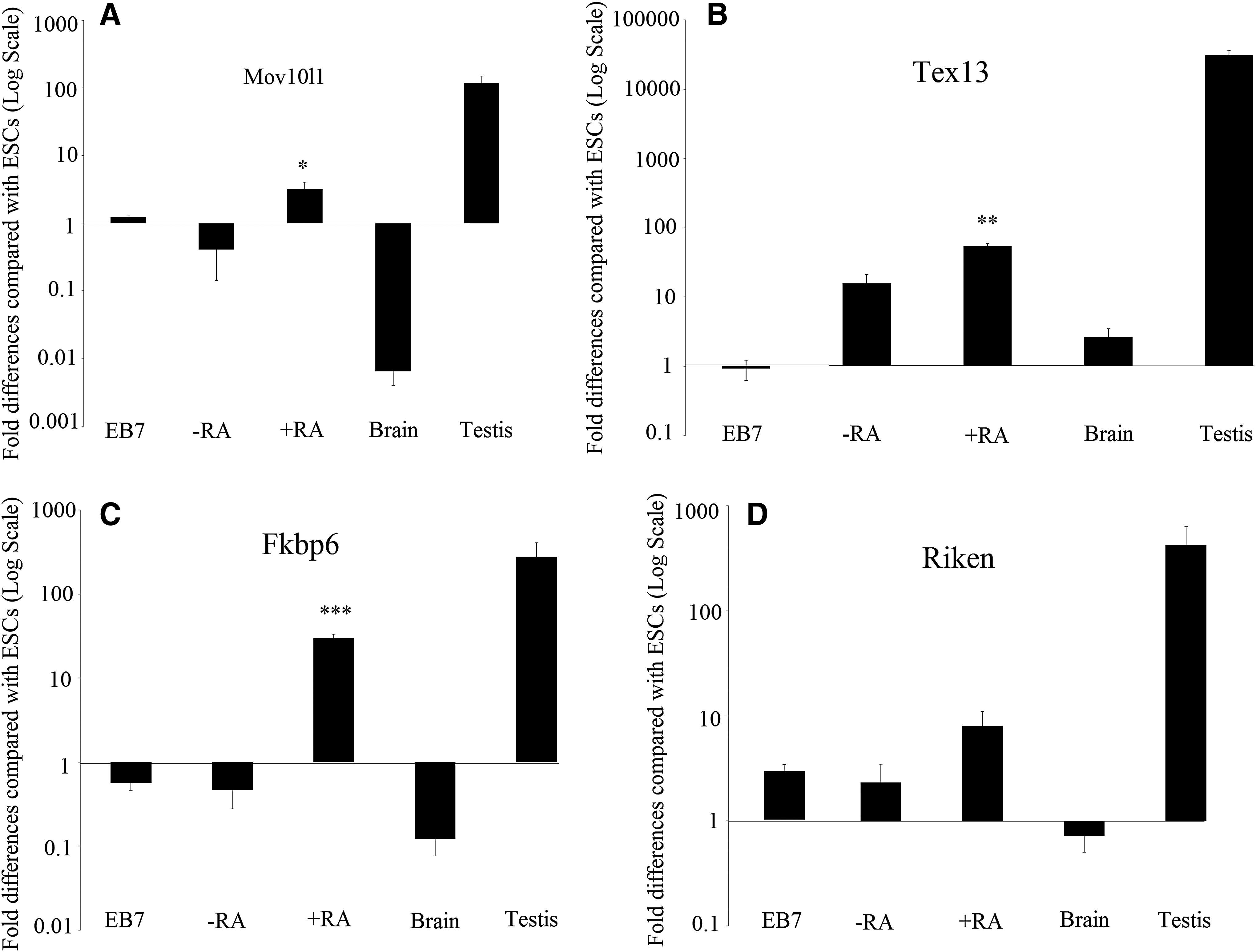
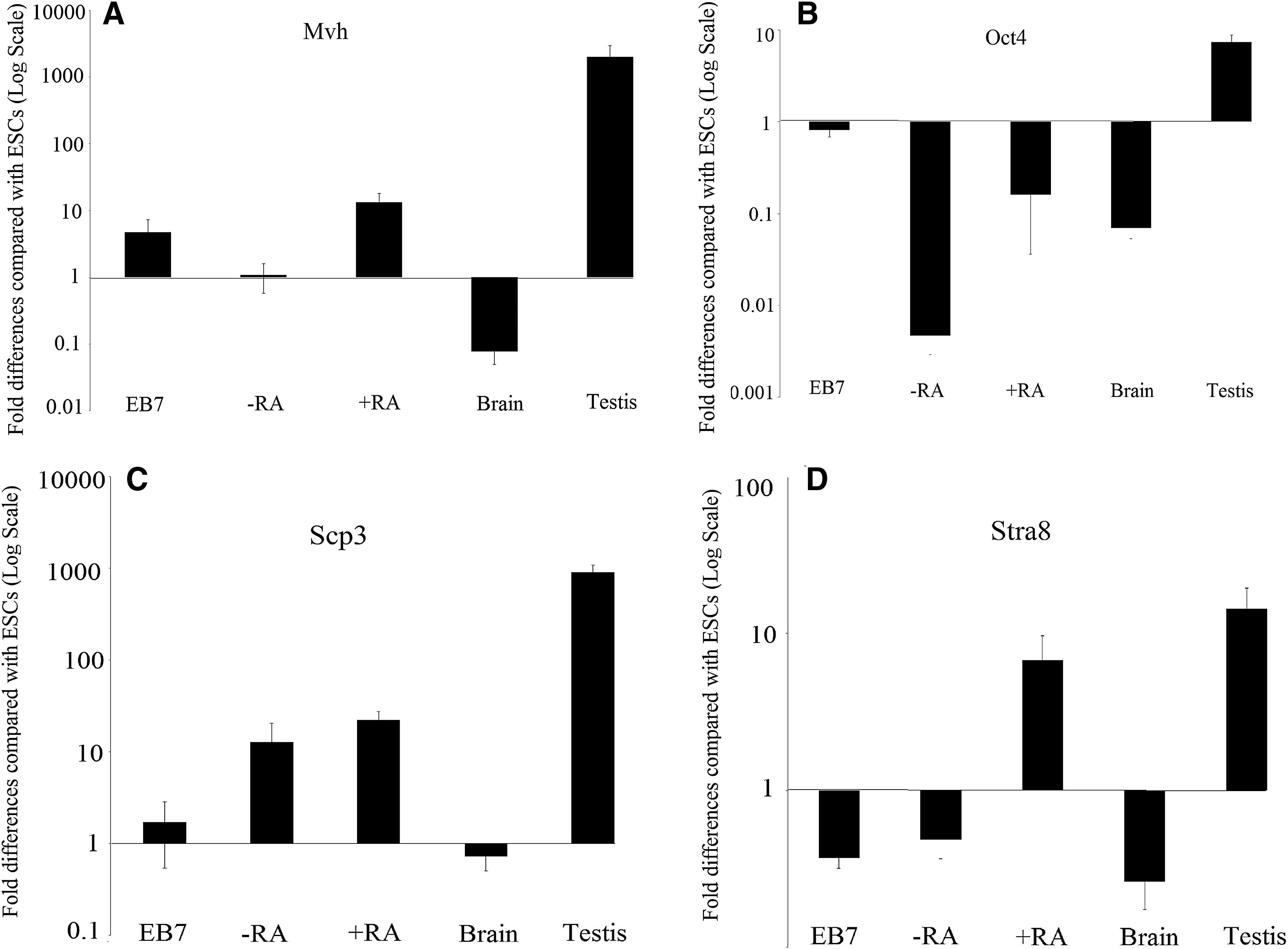
Pluripotency and known GC marker expression in ESC-derived cells was compared between three groups: (1) EB7, (2) +RA, and (3) −RA as a control group. Gene expression in the different groups revealed variations. Quantitative PCR showed that in the +RA group, expression of genes encoding Mov10l1 (p = 0.01), Fkbp6 (p = 0.00), and Tex13 (p = 0.00) were upregulated and are significantly increased compared with that of the −RA group.
Additionally, 4930432K21Rik (p = 0.1), Mvh (p = 0.07), Scp3 (p = 0.4), and stra8 (p = 0.07) were upregulated in the +RA group compared with that of the −RA group, but the differences were not significant (p > 0.05). Meanwhile, the expression levels of Tex13 (p = 0.1), Stra8 (p = 0.9), and Sycp3 (p = 0.3) were elevated and Mov10l1 (p = 0.5), Fkbp6 (p = 0.9), 4930432K21Rik (p = 0.9), and Mvh (p = 0.7) were decreased in the −RA group compared with that of the EB7 group. Oct4 was downregulated compared with its levels in ESCs in all study groups (p < 0.05) (Figs. 2–4).
The expression pattern of a number of GC-specific genes in study groups.
The expression pattern of a number of GC-specific genes in study groups.
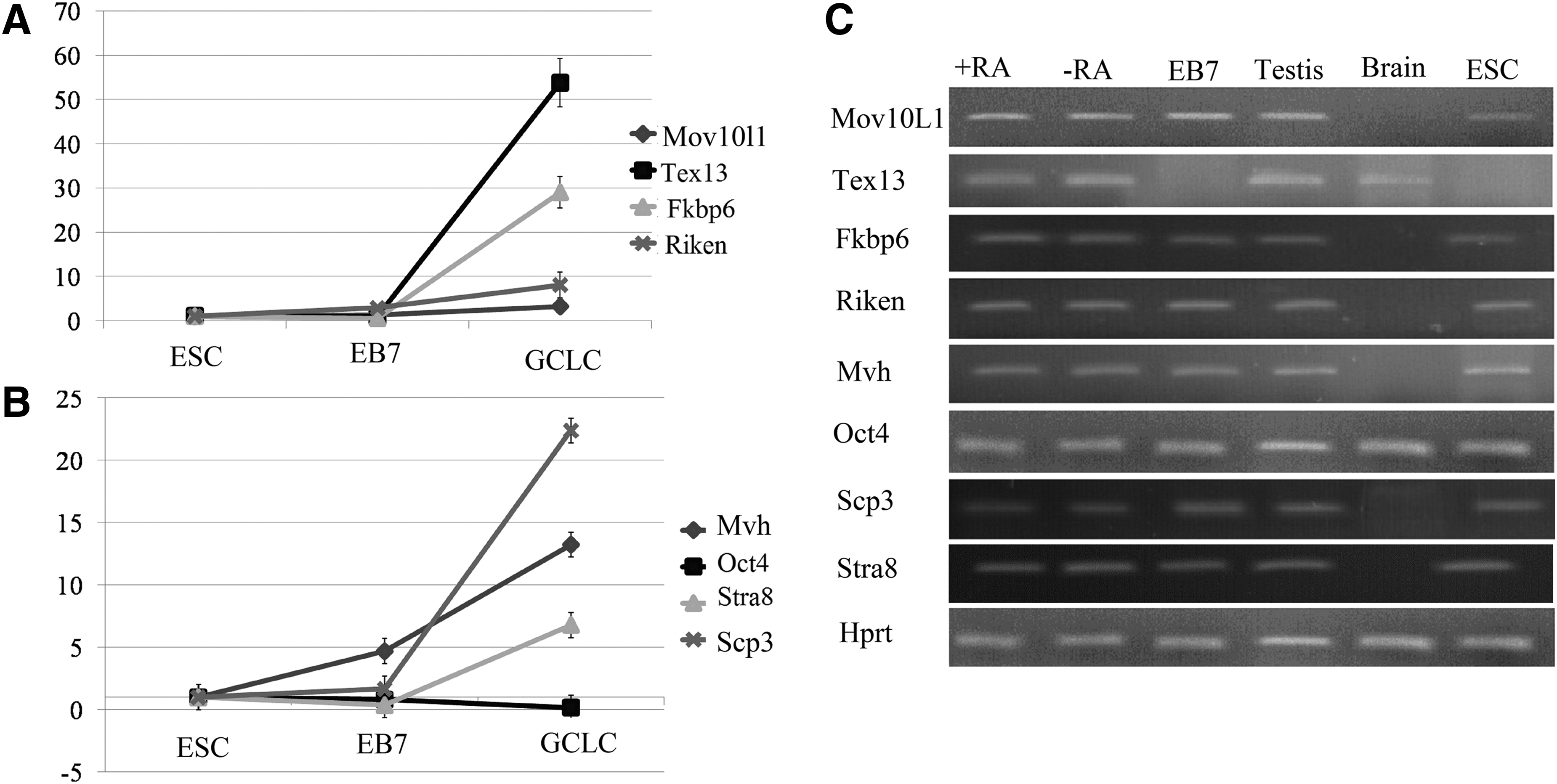
Comparison of expression level of GC-specific genes in each study groups.
Expression of GC-specific genes in in vitro-derived PGCs
We analyzed four GC-related genes (Fkbp6, Mov10l1, 4930432K21Rik, Tex13) in ESC-derived GCs. All GC-specific genes (except Mov10l1) were found to be expressed at moderate-to-high levels in PGCs. Fkbp6, Mov10l1, 4930432K21Rik, and Tex13 exhibited very low to no expression in tissues from the brain. In addition, Fkbp6, Mov10l1, Tex13, and 4930432K21Rik were expressed at moderate-to-high levels in adult testis.
We found that Tex13 mRNA was expressed in higher levels than other genes in in vitro-derived GC-like cells (GCLCs). It was ∼17-fold higher than that of Mov10L1, 6.6-fold higher than that of 4930432K21Rik, and 1.8-fold higher than that of Fkbp6.
For immunofluorescence staining, we cultured singled EBs in a chamber slide gelatinized tissue culture. After 7 days, the cells were stained with Mvh primary antibody and secondary antibody (goat anti-rabbit IgG-PE), and the expression of the Mvh protein was examined with a microscope and is shown as a red color (Fig. 5). The results showed that higher Mvh expression staining was observed in the +RA group compared with that of the −RA group, which confirmed the data from the q-RT-PCR. The cells characterizing GCLCs with round nuclei are shown by Hoechst nuclei counterstaining.
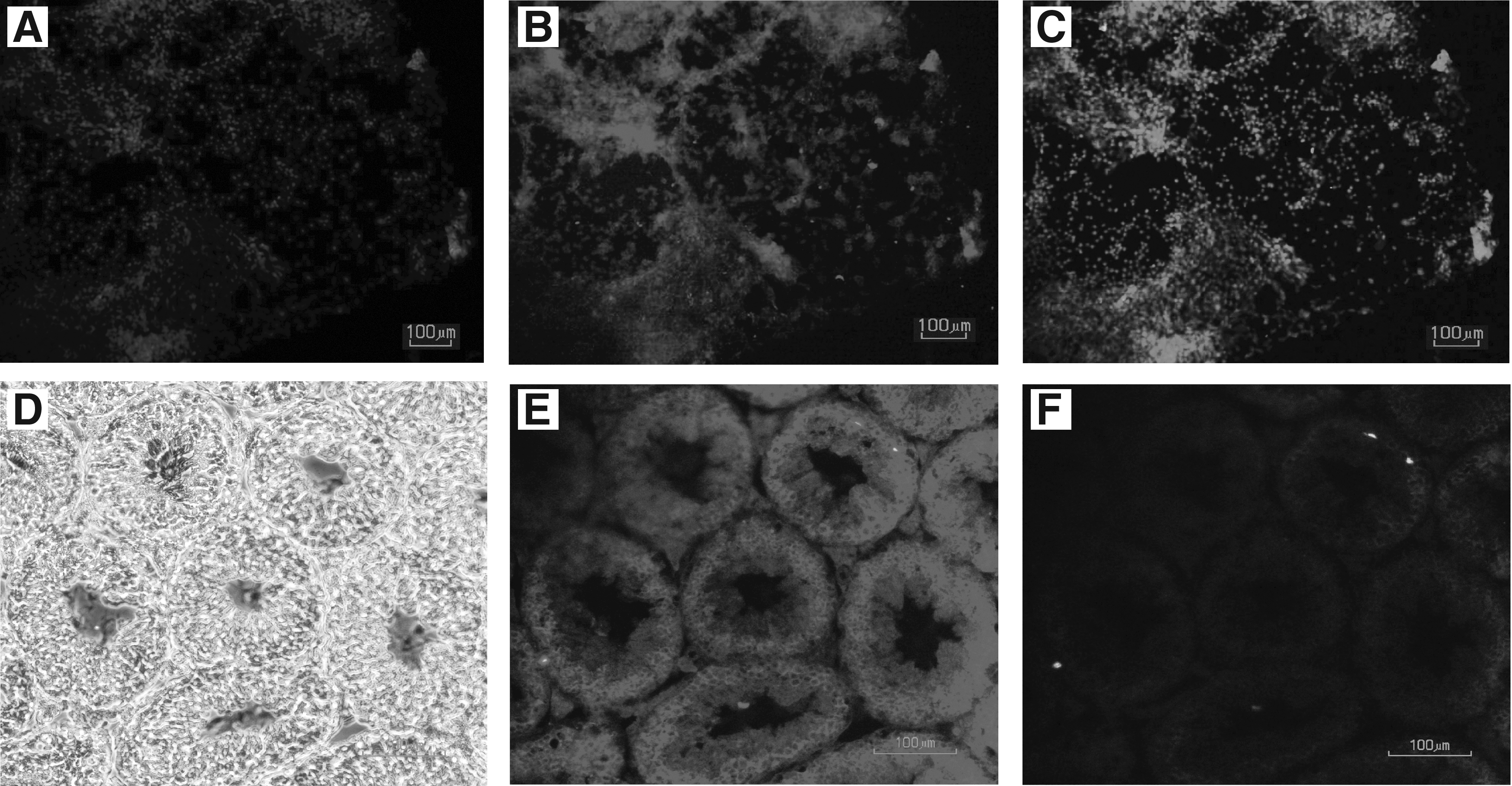
Immunofluorescence staining in study groups.
Flow cytometry
Since RA has been shown to increase the expression of GC markers by q-RT-PCR, we investigated protein expression of Mvh by flow cytometry. More Mvh-positive cells were observed in the +RA group compared with that observed in the undifferentiated ESC, −RA, and EB7 groups (p = 0.00, p = 0.00, p = 0.00, respectively). Additionally, there is a significant difference between Mhv expression in EB7 and undifferentiated ESC (p = 0.00). However, no significant differences were observed in Mvh expression on the surface of −RA cells in comparison with EB7 (p = 0.09) (Fig. 6).
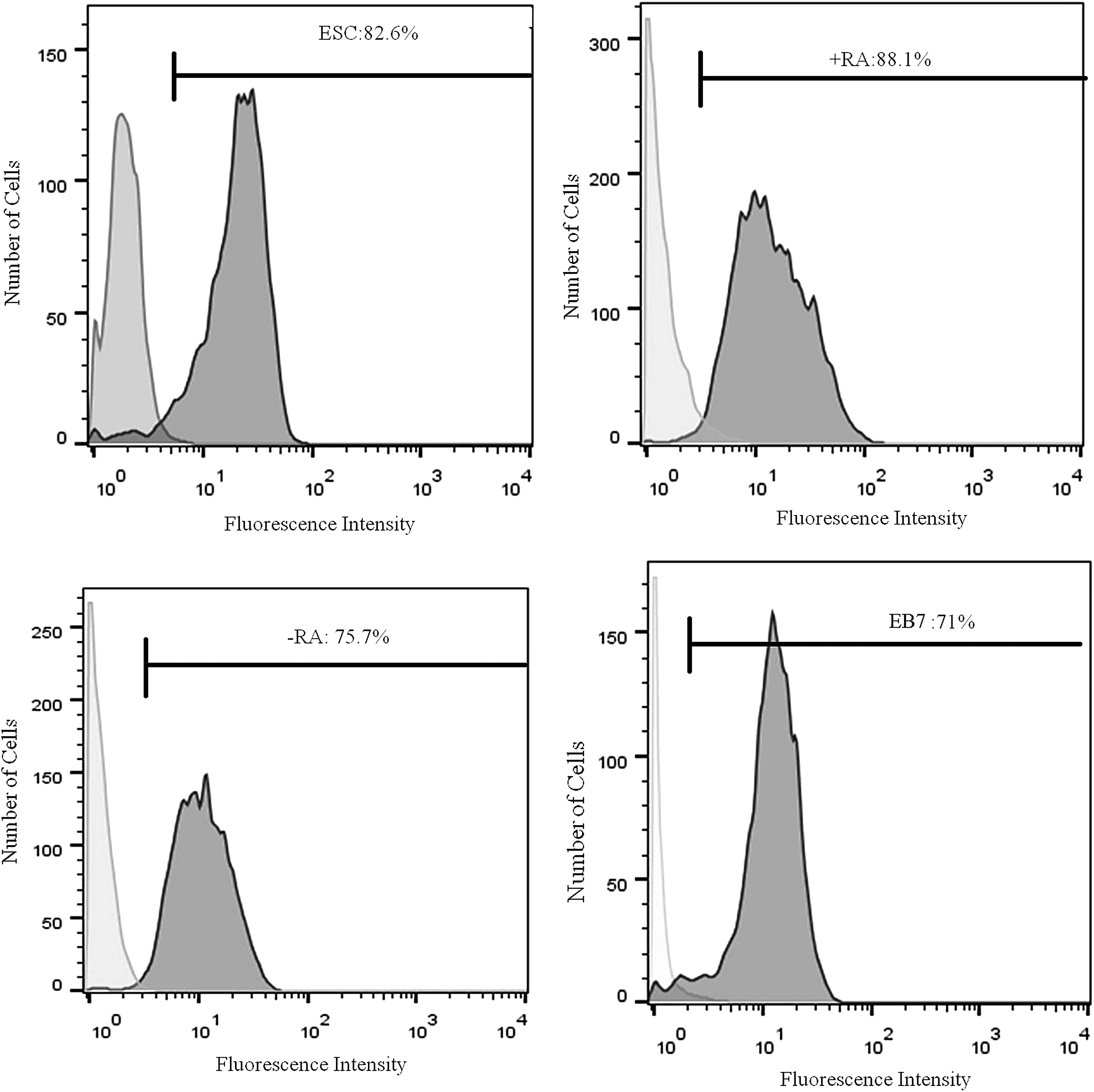
Flow cytometric analyses in study groups. Flow cytometric analyses show the expression of Mvh protein in the ESC, +RA, −RA, and EB7 cells. Undifferentiated ESC and ESC-derived GCLCs at day 7 of culture with 2 μM RA (+RA); Culture of 7-day EB without RA for 7 days control group (−RA); ESC differentiation after 7 days in suspension culture as EB (EB7).
Discussion
This study demonstrates that GC-specific genes (Fkbp6, Mov10l1, 4930432K21Rik, and Tex13) exhibit differential gene expression profiles in ESCs, GCLCs, and somatic tissues, which made them ideal and specific markers for detection of GCs during the in vitro differentiation of pluripotent stem cells into GCs. We showed that RA is a strong and effective inducer of ESC-derived GCLC. Using the expression of specific markers of GCs that are not expressed or expressed at low levels in ESCs, we can track the formation and differentiation of GCs in vitro. RA stimulates cell proliferation and prevents apoptosis (Koshimizu and Nakatsuji, 1995) and induces meiosis in migratory and postmigratory murine GCs (Zhou et al., 2008). In this study, the expression of the Mov10l1 gene was increased in RA-treated cells (+RA).
There was a significant difference in expression between +RA and that of the control group (−RA). The transcriptional level of this gene in spermatogenic cells is abundant, but in somatic cells is low. In a 15-day-old male mouse and a 7-week-old adult mouse, the expression of the Mov10l1 gene is very common in the pachytene spermatocyte. Also, after 21 postnatal days (dpp), the frequency of its transcription is continuously reduced, coinciding with the emergence of the first generation of rounded spermatozoa (Frost et al., 2010). The expression of this gene increases in the leptotene and zygotene stages of meiosis and decreases at the end of meiosis (Wang et al., 2005).
The results of this study showed that the expression of the Tex13 was significantly higher in +RA than in the control group. The results are consistent with the results of the study by Sabour et al., showing that this gene was expressed in the embryo of the 12.5 dpp male mice, as well as in the testicular tissue and sperm cells (Sabour et al., 2011). Tex13 is an X link gene that is expressed in the early stages of spermatocyte during the leptotene and zygotene stages of meiosis (Wang et al., 2005); however, it appears to undergo translational suppression before late meiosis. Tex13 was not detected in testis lacking GCs, suggesting GC-specific expression (Frost et al., 2010).
Our results showed that expression of the Fkbp6 gene in +RA was significantly increased compared with that of −RA. Mouse Fkbp6 is not involved in the initiation of synapsis, but it does play a role in monitoring progression and/or maintaining synapsis between homologous pairs. Accordingly, Fkbp6 is thought to be a component of the synaptonemal complex essential for sex-specific fertility, and the commitment of homologous chromosome pairing in meiosis (Ikadai et al., 1992). The expression of Fkbp6 in male mice at 12.5 dpp and testicular tissue of adult mice is high, but decreases significantly in sperm, indicating that its main role is in the creation of a synaptic complex in the paired homologous chromosome (Sabour et al., 2011).
In this study, 4930432K21Rik expression level was increased in the +RA group compared with that of the −RA, but there was no statistically significant difference. 4930432K21Rik is a new gene with unknown function. This gene is expressed in PGCs of mouse embryos, mice adult testis, spermatozoa, and oocytes. C19ORF57 is the human equivalent of 4930432K21Rik that expressed in the lower level in testis than other three genes (Fkbp6, Mov10l1, and Tex13) (Wang et al., 2005). There are not many reports on this gene.
In this study, in addition to studying four GC-specific genes, four other genes that are expressed in GCs were also studied. The expression levels of Mvh, a sex-linked gene, in ESCs treated with +RA were increased compared with that of −RA. The expression of this gene increases with the onset of meiosis and remains high until the end of spermatogenesis (Wang et al., 2005). The absence of the Mvh results in halting of the zygotene stage in male gametes (Tanaka et al., 2000). In this study, Mvh expression at the protein level as analyzed by immunocytochemistry and flow cytometry showed a greater increase in the +RA group than in the control group, which confirmed the findings of RT-PCR. Mvh is a cytoplasmic protein and is a product of the Vasa homolog gene, which is induced by somatic cell of the genital crest, and remains until the formation of postmeiotic GCs.
It has also been shown through immunohistochemistry that the Mvh protein in PGCs was expressed exclusively after embryonic gonad colonization at E12.5 (Anderson et al., 2007) and in GCs undergoing gametogenesis until the postmeiosis stage in each males and females (Toyooka et al., 2000). In Mvh knockout mice, female mice have a normal oogenesis period, whereas male mice are infertile. This suggests halting of meiosis in the zygote stage (Anderson et al., 2007).
In this study, Oct4 gene expression was decreased in all studied groups compared with ESCs. Oct4 expression increases in pluripotent cells, such as ESCs, and decreases after differentiation. During gastrolization and stem cell differentiation, Oct4 expression decreases and is limited to ESCs (Yeom et al., 1996). Depending on Oct4 characteristics, it is expressed in ESCs, ECs, and embryonic GCs (O'shea, 2004.). In vivo studies have shown that GCs exhibit a high expression of Oct4 until E13.5, then decreases in the zygote/pachytene stages of first meiosis around E16.5 (Pesce et al., 1998).
In our study, the expression of Sycp3 in the +RA group was higher than that of the −RA group. This gene is essential for synaptonemal complexes, chromosomal synapse, and male fertility. Male mice lacking Sycp3 have defects in the formation of axial/lateral elements, and chromosomes lacking this gene are not able to form synaptic complexes (Yuan et al., 2000). This event causes the Sycp3−/− mice to stop at the zygote stage (Yang et al., 2006). The expression of Sycp3 begins at the primary spermatocyte and ceases at the spermatid stage (Aarabi et al., 2006). We concluded that the expression of the Stra8 gene increased in the +RA group and decreased in the −RA and EB7 groups. RA, which stimulates Stra8 expression, has an inductive role in the onset of meiosis (Koubova et al., 2006).
Recent studies have shown that RA signals play an important role in regulating the male GC cycle (Spiller and Koopman, 2011). RA prevents mitotic arrest in the XY gonad at E11.5 to E13.5. This increases the Stra8 concentration, which triggers the onset of meiosis at E11.5 in vitro (Koubova et al., 2006). Interestingly, RA increases the expression of Stra8 in nondifferentiated spermatogonia, but has a low effect on differentiated spermatogonia (Oulad-Abdelghani et al., 1996). Initially, the expression of this gene is seen in immature testis and GCs with mitotic activity after birth, and then expression is increased in the adult testis in undifferentiated GCs (Wang et al., 2016).
Conclusion
These four specific GCs genes are expressed in the testis, but not the ovary, which indicates their role in the development of male gametes. According to the findings, downregulation of Oct4 as a pluripotency factor as well as the expression of meiosis markers, the hypothesis is raised that ESCs are differentiated by RA, and have been introduced into the zygote/pachytene of first meiosis as GCLCs. So induction of mESCs by 2 μM RA may cause differentiation toward GCLCs. Improving in vitro GC differentiation with high efficiency may simplify the generation of mature gametes for understanding of biology of gametogenesis.
Footnotes
Acknowledgments
The authors are grateful to the Vice Chancellor for Research and Technology of the Babol University of Medical Sciences, the Health Research Institute and the staff of the Stem Cell Laboratory of the Reproductive Health and Infertility Research Center of Fatemeh Al Zahra Hospital, and the Cellular and Molecular Research Center. The project was sponsored by the research grant, number 9337828.
Authors' Contributions
M.G.T.: designed and performed experiments, analyzed data, and co-wrote the article; S.G.A.J.: supervised the research, designed experiments, and co-wrote the article; M.G.H.: designed experiments and performed transporter experiments; A.A.A.: coworker for performing experiments. Neda Mahdinejad: coworker for performing experiments.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.