
Abstract
Abstract
Ectrodactyly-Ectodermal dysplasia-Clefting (EEC) syndrome is a rare monogenic disease with autosomal dominant inheritance caused by mutations in the TP63 gene, leading to progressive corneal keratinocyte loss, limbal stem cell deficiency (LSCD), and eventually blindness. Currently, there is no treatment available to cure or slow down the keratinocyte loss. Human oral mucosal epithelial stem cells (hOMESCs), which are a mixed population of keratinocyte precursor stem cells, are used as source of autologous tissue for treatment of bilateral LSCD. However, hOMESCs from EEC patients have a reduced life span due to TP63 mutations and cannot be used for autologous transplantation. Human induced pluripotent stem cells (hiPSCs) represent a potentially unlimited source of autologous limbal stem cell for EEC patients and can be genetically modified by genome editing technologies to correct the disease ex vivo before transplantation. In this study, we describe for the first time the generation of integration-free EEC-hiPSCs from hOMESCs of EEC patients by Sendai virus vector and episomal vector-based reprogramming. The generated hiPSC clones expressed pluripotency markers and were successfully differentiated into derivatives of the three germ layers, as well as toward corneal epithelium. These cells may be used for EEC disease modeling and open perspectives for applications in cell therapy of LSCD.
Introduction
E
The idea of using these cells to build surrogate corneal sheets comes from the observation that these cells share many features with limbal stem cells (Nishida et al., 2004) and can be easily grown in cell culture, without undergoing keratinization (Hata et al., 1995). Unfortunately, the limited life span and early differentiation of hOMESCs derived from EEC patients hamper ex vivo expansion, thus impeding successful gene correction approaches (Barbaro et al., 2016). Human induced pluripotent stem cells (hiPSCs) (Takahashi and Yamanaka, 2006) have been applied to several fields of study, such as drug discovery, regenerative medicine, and disease modeling (De Vos et al., 2016; Trevisan et al., 2015).
Recently, the results of the first clinical research using autologous retinal epithelial cells derived from hiPSCs in age-related macular degeneration showed the feasibility to cure ocular diseases using hiPSC-derived cells (Mandai et al., 2017). In the case of a genetic disease such as EEC, these cells can be genetically modified to correct the mutation before autologous transplantation. So far, EEC-hiPSCs have been generated from dermal fibroblasts using lentiviral vectors, which pose the risk of insertional mutagenesis (Shalom-Feuerstein et al., 2013). For clinical applications, safer and integration-free reprogramming protocols are advisable.
Furthermore, other cell sources, such as hOMESCs obtained through oral biopsies, could provide better results for cell therapy of ocular defects. hOMESCs are composed by a mixed population of keratinocyte precursors, which can be more efficiently reprogrammed to iPSCs than fully differentiated fibroblasts and keratinocytes, and share the same ectodermal origin as those of the cornea (Burillon et al., 2012; Chen et al., 2009; Nishida et al., 2004), which represents an advantage for differentiation toward corneal epithelium (Bar-Nur et al., 2011; Kim et al., 2010). Among nonintegrative reprogramming approaches, a very efficient method is represented by Sendai virus vectors (SeVs), which can transduce a variety of cell types without entering the nucleus and are diluted from the cells with the physiological cell division (Fusaki et al., 2009).
Other nonintegrative methods are based on episomal vectors that have the advantage of replicating without integrating into the host genome and to be eliminated with cell division (Nanbo et al., 2007; Okita et al., 2011). Episomal vectors are widely used for reprogramming strategies, since they are easy to produce; only a single transfection event is sufficient for pluripotency induction in target cells and many cell types can be transfected, including keratinocyte-like cells. Unfortunately, hOMESCs are particularly recalcitrant to liposome-based transfection (Ino et al., 2008).
In the present study, we applied SeVs, as well as a new protocol exploiting episomal vectors coupled to nucleofection technology, to generate integration-free hiPSCs by reprogramming of hOMESCs from a healthy subject and two EEC patients carrying severe TP63 mutations (i.e., R279H and R304Q) (Barbaro et al., 2016). hOMESC-derived hiPSCs were successfully differentiated toward corneal epithelium. Characterization results of representative hiPSC clones derived from the two EEC patients (R279H-hOMESCs; R304Q-hOMESCs) and from the healthy control (wt-hOMESCs) are reported elsewhere (Alvisi et al., 2018b; Trevisan et al., 2018a, 2018b).
Materials and Methods
Study participants
A healthy subject and two EEC patients carrying either the R279H or the R304Q mutations, respectively, in the TP63 gene were included in this study. The study was approved by the Venetian Ethics Committee for Clinical Research Studies (Prot. 2009/77661, 19th November 2009). All patients provided written informed consent to participate in the study.
Cell culture and media
3T3-J2 fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% GlutaMAX™, and 1% penicillin/streptomycin (all from Gibco; Thermo Fisher Scientific, Waltham, MA) and irradiated at 60.00 Gy. Irradiated mouse embryonic fibroblasts (MEFs, MTI-GlobalStem; Thermo Fisher Scientific) were cultivated with the same medium and were seeded at a density of 2 × 103 cell per cm2. hOMESCs and human limbal epithelial stem cells (h-LESCs) were isolated from fresh oral mucosal and limbal biopsies of study participants, respectively.
Cells were grown in keratinocyte growth medium (KGM) as previously described (Barbaro et al., 2016). hiPSCs were grown either on MEF feeder layer in human embryonic stem (hES) medium or on Matrigel (BD Biosciences, Franklin Lakes, NJ) in mTeSR1 (STEMCELL Technologies, Vancouver, BC, Canada). hES medium consisted of DMEM-F12 supplemented by 20% knockout serum replacement, 1% nonessential amino acids (NEAA), 1% GlutaMAX, 1% penicillin/streptomycin, 0.1% β-mercaptoethanol (all from Gibco; Thermo Fisher Scientific), and basic fibroblast growth factor, b-FGF, 10 ng/mL (ORF Genetics, Kopavogur, Iceland).
Establishment of optimal nucleofection conditions for hOMESCs
hOMESCs at passage 2 were seeded on 3T3-J2 precoated 24-well plates and maintained in KGM at 37°C in a 5% CO2, 95% humidified air incubator. The following day 10 ng/mL epidermal growth factor, EGF (ORF Genetics), was added to media. One week later, cells reached 80% confluency and were processed for transfection with a maxGFP encoding plasmid (pMAX) in combination with a 4D-Nucleofector™ system, equipped with a Y unit and 16-well Nucleocuvette™ Strips (Lonza, Basel, Switzerland). To this end, cells were washed twice in PBS, treated for 8 minutes with trypsin (Gibco, Thermo Fisher Scientific, 1 mL/well), and resuspended in DMEM (10% FBS) at a final concentration of 2 × 106 cells/mL.
Cells were then subjected to nucleofection following either the Amaxa 4D-Nucleofector basic protocol for human stem cells in combination with two Primary Cell 4D-Nucleofector® X Kits (P3 and P4 Primary Cell Kits) or the Amaxa 4D-Nucleofector basic protocol for primary mammalian epithelial cells in combination with the P4 Primary Cell 4D-Nucleofector X Kit. Overall, 19 different conditions were tested. Each reaction was performed using 2 × 105 cells in the presence of 20 μL of Nucleofection solution and 400 ng of pMAX. Upon nucleofection, 80 μL of KGM was immediately added to the cells that were then moved to 3T3-J2 precoated 24-well plates containing 12 mm glass coverslips, in a final volume of KGM of 1 mL. Twenty-four hours later, medium was changed to remove cell debris using KGM.
Confocal laser scanning microscopy analysis of transgene expression
Forty-eight hours after transfection, cells were incubated for 30 minutes with DRAQ5 (5 μM; Thermo Fisher Scientific) to stain cell nuclei. Cells were washed twice in PBS, fixed with 4% paraformaldehyde (Sigma Aldrich, St. Louis, MO) containing 2% saccharose for 15 minutes at room temperature, before being mounted onto glass coverslips with Fluoromount-G (Southern Biotech, Birmingham, AL). Samples were processed by confocal laser scanning microscopy (CLSM) using a NIKON A1 system, equipped with a 10 × objective (Nikon, Amsterdam, Netherlands).
For each condition, two randomly chosen fields were acquired after excitation of the samples at 488 and 633 nm, to detect images relative to the GFP and DRAQ5 channels, respectively. The number of positive cells was determined using the NIH ImageJ 1.62 public domain software. Data were analyzed and plotted using Prism 6 (GraphPad, La Jolla, CA) software as described previously (Alvisi et al., 2018a).
Reprogramming of hOMESCs into hiPSCs
R279H- and R304Q-hOMESCs at passage 2, growing on 3T3-J2 feeder layers in KGM, were detached by three cycles of trypsinization for 8 minutes at 37°C. For episomal vector reprogramming, 5 × 105 cells were centrifuged at 1100 rpm for 5 minutes, washed once with PBS, and then resuspended in 100 μL of P3 solution (Lonza). Two micrograms of each of the three plasmids carrying the reprogramming factors (pCXLE-hOCT3/4-shp53-F, pCXLE-hSK, pCXLE-hUL—a gift from Shinya Yamanaka, Addgene, Cambridge, MA; plasmids # 27077, 27078, and 27080, respectively) were added, and the cells were nucleofected using program EL110.
Upon nucleofection, 500 μL of KGM was added to the suspension, and cells were allowed to recover at 37°C for 5 minutes. For SeV reprogramming 2.5 × 105 hOMESCs at passage 2 were transduced with the SeVs bearing Yamanaka's factors OCT4, SOX2, KLF4, and cMyc at a multiplicity of infection of three using the CytoTune®-iPS Sendai Reprogramming Kit (Life Technologies) following the manufacturer's protocol. Nucleofected and transduced hOMESCs were then seeded on dishes previously coated with MEFs in KGM. From the day after the medium was changed every other day until day 5, the medium was switched to a mixture of KGM and hES medium at a ratio 1:1.
Forty-eight hours later, the medium was switched to hES medium and was changed daily. MEFs were added weekly to guarantee the feeder layer contribution to the reprogramming. Once emerged, hiPSC colonies were manually picked and moved to six well plates previously coated with MEFs and expanded for further characterization.
Quantitative analysis of OCT4 expression in hOMESC-derived hiPSCs by flow cytometry
hiPSCs growing on Matrigel in mTeSR1 were harvested by treatment with Accutase (STEMCELL Technologies), fixed with paraformaldehyde 4% for 10 minutes at 37°C, and then treated with 90% cold methanol/PBS overnight at 4°C. The next day, primary antibody (Goat anti-OCT3/4 [N-9], sc-8628, Santa Cruz Biotechnology, Inc., Dallas, Texas) was added to the cells and incubated for 1 hour at reverse transcriptase (RT), followed by incubation with the secondary antibody (Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, A-11055; Invitrogen, Thermo Fisher Scientific) for 1 hour at room temperature. Flow cytometry was performed in a BD LSR II Cytofluorimeter (BD Biosciences, San Josè, CA), and data were analyzed with the Flowing software 2.5.1 (http://flowingsoftware.btk.fi/)
Analysis of pluripotency marker expression on hOMESC-derived hiPSCs by RT-PCR
hiPSCs grown on Matrigel in mTeSR1 were harvested by treatment with Accutase, and total RNA was purified with the RNeasy Mini Kit (Qiagen, Hilden, Germany), following the manufacturer's instructions. Samples were further treated with Turbo DNase (Ambion, Thermo Fisher Scientific) for 30 minutes at 37°C. Total RNA was quantified by NanoDrop spectrophotometer (NanoDrop, Thermo Fisher Scientific), and 1 μg was reverse transcribed into cDNA. For each reaction, a reverse transcription-negative control reaction was performed adding all the reagents except for the RT enzyme. cDNAs were used as a template for the RT-PCR amplifying several pluripotency associated genes, such as DNMT3b, TERT, NANOG, and REX1. ACTIN was amplified as the control housekeeping gene.
Analysis of pluripotency marker expression on hOMESC-derived hiPSCs by indirect immunofluorescence
hiPSCs grown on MEFs in hES medium were treated with Collagenase IV (Thermo Fisher Scientific), scraped and seeded into 24-well plates containing 12 mm glass coverslips coated with MEFs. Five days after passaging, cells were washed thrice with PBS and fixed in paraformaldehyde 4% for 20 minutes at room temperature. Fixed cells were then permeabilized with PBS/0.1% Triton X-100 (Sigma Aldrich) for 15 minutes at room temperature. Unspecific binding sites were saturated using PBS/4% BSA (Sigma Aldrich) for 1 hour at room temperature, after which primary antibodies diluted in PBS/4% bovine serum albumin (BSA) were added to the cells and incubated overnight at 4°C.
After three washes with PBS/0.05% Tween 20 (Sigma Aldrich), cells were subsequently incubated with Alexa Fluor® 488 secondary antibodies (Thermo Fisher Scientific) diluted in PBS for 1 hour at room temperature. Cell nuclei were stained by incubation for 5 minutes with DAPI 300 nM (Thermo Fisher Scientific) in PBS followed by three washes with PBS. Coverslips were observed using an inverted fluorescent microscope (DFC 420C; Leica, Wetzlar, Germany), and digital images were acquired with LAS V3.8 software and processed with NIH ImageJ 1.62 public domain software as described previously (Avanzi et al., 2013). The following primary antibodies were used: 1:200 Oct3/4 (N-9), 1:200 GKLF (H-180), (both from Santa Cruz Biotechnology), and 1:50 SSEA4 (Abcam, Cambridge, UK).
Embryoid body formation test
hiPSCs growing on MEFs in hES medium supplemented with b-FGF were detached with collagenase IV treatment combined with mechanical scraping. Cells were forced to grow for 7 days in suspension into ultralow density attachment plates (Corning, NY) in hES medium depleted of b-FGF, and medium was changed every 3 days.
After 1 week, the formed embryoid bodies (EBs) were moved to a six well plate previously coated with 0.1% porcine gelatin (Merck Millipore, Billerica, MA) and allowed to grow in adhesion for a further week in DMEM (10% FBS). Cells were then detached by trypsin treatment, and total RNA was extracted and reverse transcribed as above described. cDNAs were then used as templates for RT-PCR amplifying markers of the three germ layers, ectoderm (TUBB and PAX6), mesoderm (FLK1 and PECAM), and endoderm (AFP and GATA4). ACTIN was amplified as housekeeping gene. For each RT-PCR, a RT sample was included.
Differentiation of hiPSCs into corneal keratinocytes
hiPSCs were differentiated into corneal keratinocytes as previously described, with slight modifications (Mikhailova et al., 2014). Briefly, hiPSCs growing on MEFs in hES medium were detached and allowed to grow for 4 days in suspension in hES medium supplemented with 10 μM SB-505124 (Sigma-Aldrich), 10 μM IWP-2 (Merck Millipore), and 50 ng/mL b-FGF with daily medium change. Cells were then seeded on 3T3-J2 fibroblasts and allowed to grow for 30 days in KGM. At different time points, cells were tested by quantitative real-time RT-PCR or immunofluorescence (IIF) for the expression of the markers typical of the corneal stage, that is, ΔNp63α, K12, ABCB5, pan-cytokeratins.
Statistical analysis
All experiments were performed thrice in triplicate; data are presented as mean ± standard deviation of the mean (SD). Comparison between groups was performed with the Student t-test, and p-values of 0.05 or less were considered significant.
Results and Discussion
Expression of pluripotency and differentiation markers in hOMESCs
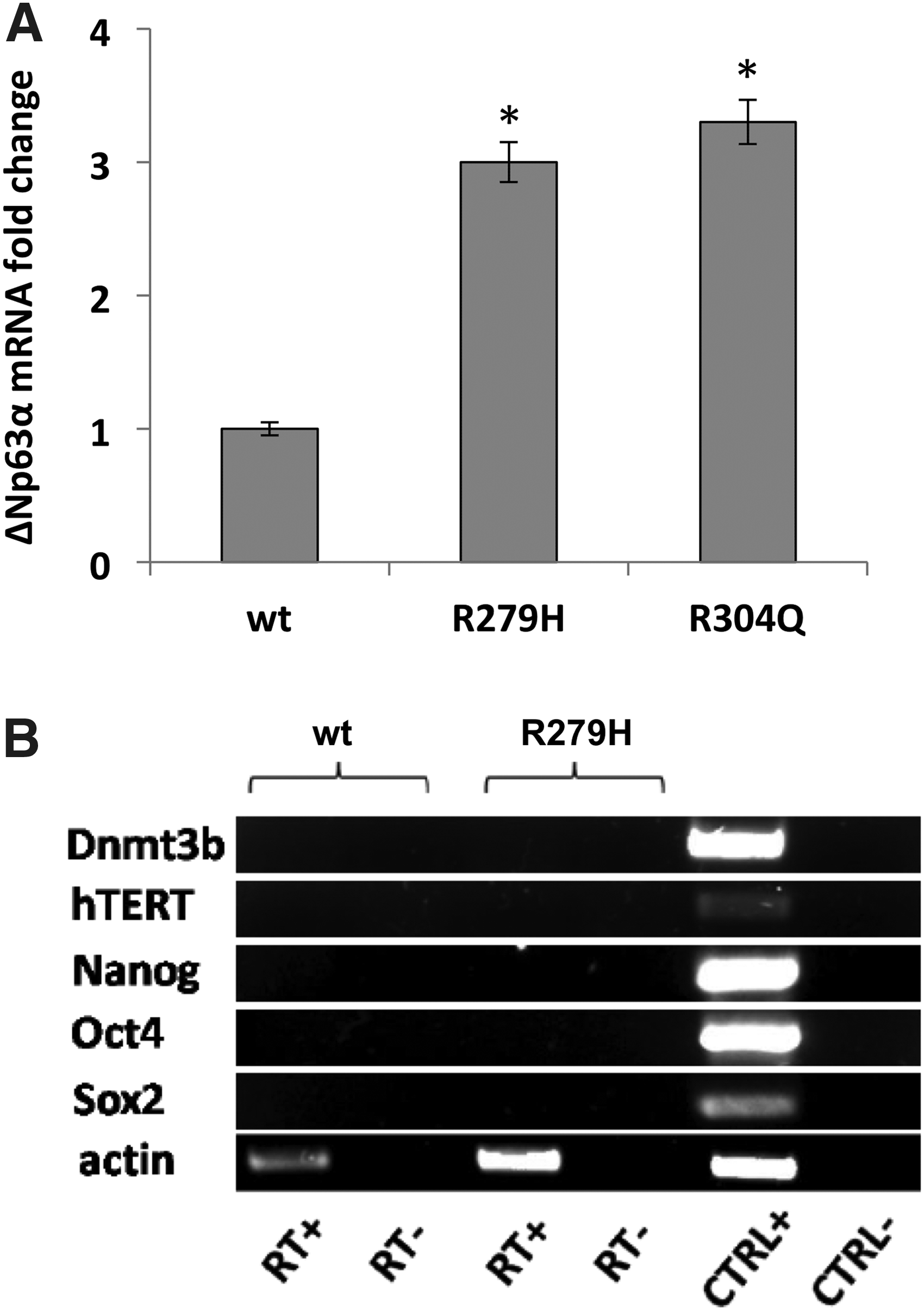
hOMESCs from a healthy donor (wt), as well as from two patients bearing the two most severe TP63 mutations known to date (R279H and R304Q), were obtained by oral biopsy. Since p63 is a key regulator of cell proliferation, survival, and apoptosis (Bergholz and Xiao, 2012) and its isoform ΔNp63 is a positive regulator of reprogramming (Alexandrova et al., 2013), we sought first to investigate the expression levels of ΔNp63α in the examined samples. Expression levels of ΔNp63α from EEC patients were threefold lower than those from the wt (Fig. 1A).
Characterization of hOMESCs:
This result is consistent with the shorter life span of EEC-hOMESCs than wt cells (Barbaro et al., 2016), due to altered expression profiles of the ΔNp63α isoform (Browne et al., 2011). Importantly, when characterized in terms of pluripotency and marker expression, wt- and EEC-hOMESCs did not show any expression of pluripotency markers such as DNMT3b, hTERT, NANOG, OCT4, and SOX2 (Fig. 1B).
Establishment of optimal nucleofection conditions for hOMESCs
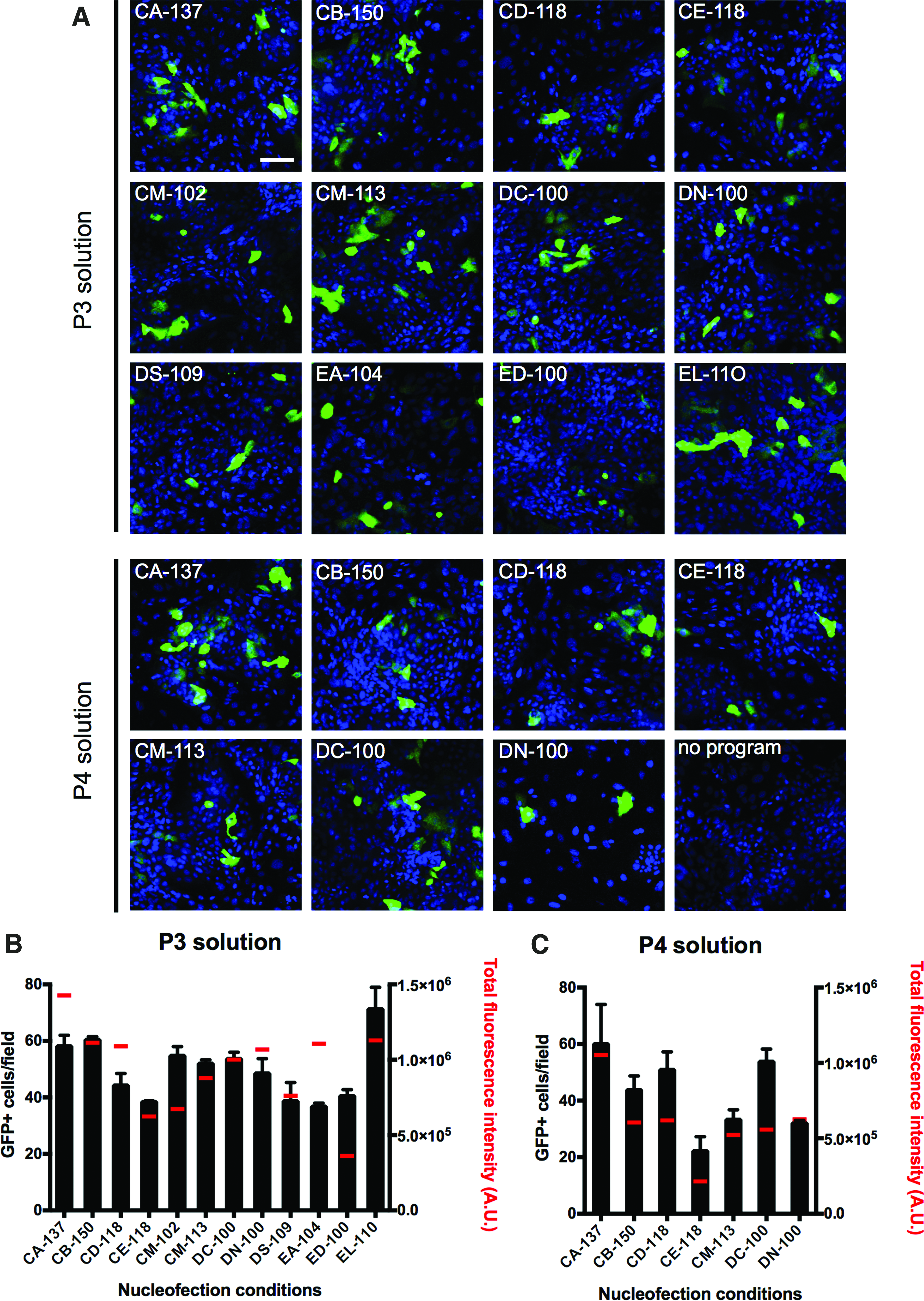
To develop a reprogramming method based on episomal vectors, we initially set up a nucleofection protocol to transfect hOMESCs. To this aim, we used a maxGFP reporter plasmid, using 19 different nucleofection conditions, which were selected based on the manufacturer's recommendations (Fig. 2A). At 48 hours post transfection, levels of transgene expression were assessed as described in the Materials and Methods section (Fig. 2A). Transfection efficiency was evaluated both by scoring the average number of GFP-positive cells and by quantifying the total fluorescence intensity (Fig. 2B, C). Our data indicate that each tested condition resulted in hOMESCs being successfully transfected, as indicated by the presence of GFP-positive cells.
Optimization of nucleofection conditions for hOMESCs.
Nucleofection using the P3 solution resulted both in a higher number of transfected cells per field (49.8 ± 10.6 vs. 42.3 ± 13.6) and in a higher fluorescence intensity [9.8 × 105 ± 2.8 × 105 arbitrary units (A.U.) vs. 6.0 × 105 ± 2.5 × 105 A.U.], than solution P4. In particular, transfection with program EL-110 in the presence of solution P3 resulted in the highest transfection efficiency (71.3 ± 11.0 positive cells per field), while transfection with program CA-137 and solution P3 resulted in detection of the highest total fluorescence (approximately 16 ± 4 × 105 A.U.). Since reprogramming efficiency mainly depends on the number of cells expressing the transgenes rather than the intensity of expression levels, program EL-110 in combination with solution P3 was chosen for hOMESC reprogramming.
Generation of hiPSCs from hOMESCs derived from a healthy donor and EEC patients
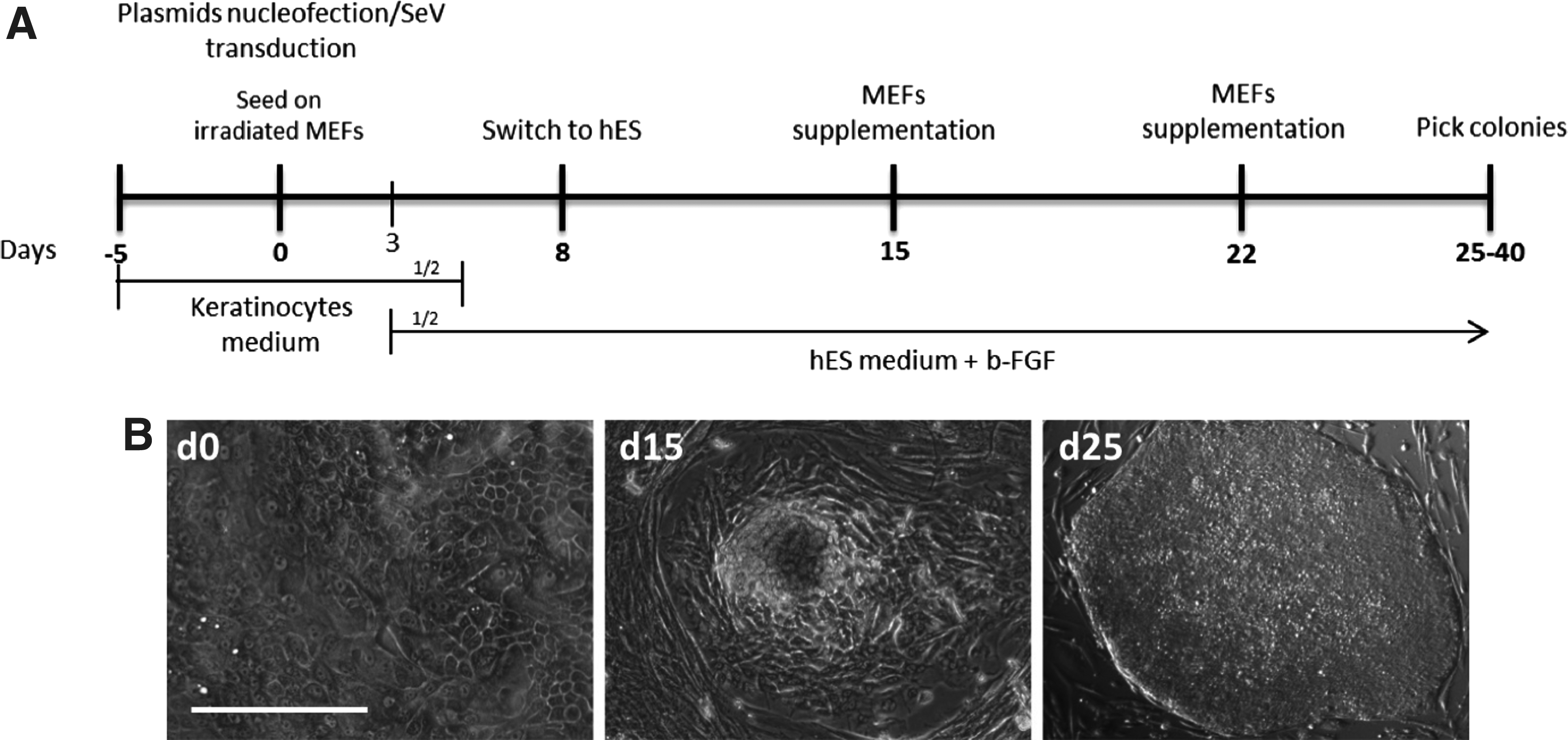
hOMESCs from a healthy donor (wt-hOMESCs) and from the two patients affected by the EEC syndrome (EEC-hOMESCs) were subjected to reprogramming using episomal vectors and SeV (Okita et al., 2011) (Fig. 3A). To adapt the cells to the new growth conditions, nucleofected and transduced hOMESCs were seeded on MEFs, and the medium was gradually switched from KGM to hES medium (Fig. 3). Morphological changes could be observed from 10–15 days post transduction/transfection (p.t.) as cells started to appear smaller in size and to have colony-like growth trends (Fig. 3B, middle panel).
Reprogramming of hOMESCs into hiPSCs.
The first hiPSC-like colonies started to emerge around 20–25 days p.t., exhibiting defined edges and being composed by compacted cells with faint cytoplasm and exposed nuclei (Fig. 3B, right panel). Reprogramming efficiency was calculated as the percentage of hiPSC-like colonies formed over the starting number of hOMESCs and ranged from 0.05% to 0.15%. The derived hiPSCs showed the absence of transgene expression and EBNA1 sequences from the episomal vector, as well as a normal karyotype. Full characterization of representative hiPSC clones derived from R279H-hOMESCs, R304Q-hOMESCs, and wt-hOMESCs is reported elsewhere (Alvisi et al., 2018b; Trevisan et al., 2018a, 2018b).
hOMESC-derived hiPSCs express pluripotency markers and can be differentiated into the derivatives of the three germ layers
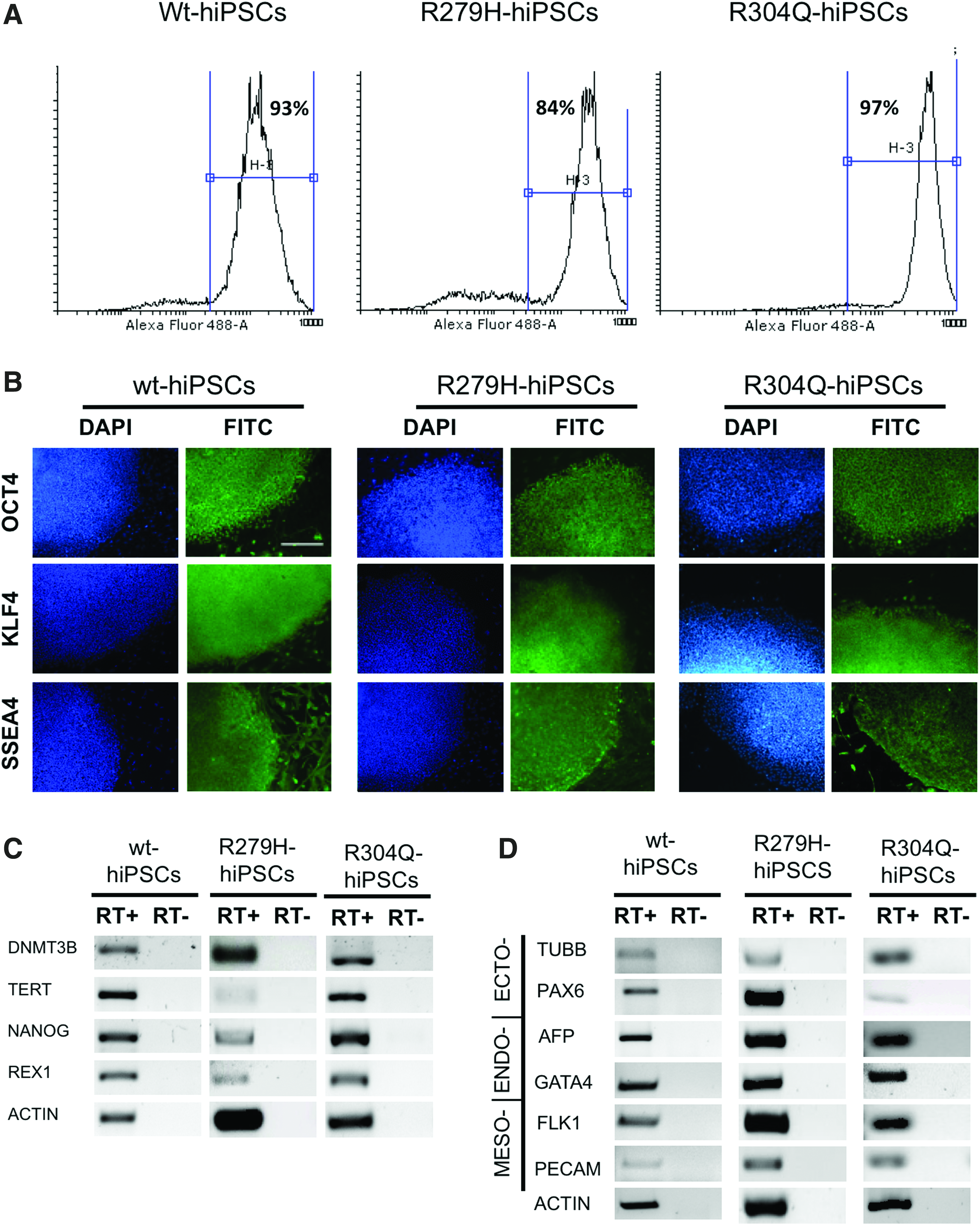
We subsequently analyzed expression of a set of pluripotency markers by flow cytometry, IIF, and RT-PCR to test the differentiation potential of wt- and EEC-hOMESC-derived hiPSCs. All cells expressed several markers of pluripotency, including OCT4, KLF4, and SSEA4, as shown by IIF and flow cytometry analysis (Fig. 4A, B), as well as DNMT3b, TERT, REX1, and NANOG, as shown by RT-PCR (Fig. 4C), indicating that they represented bona fide pluripotent stem cells.
Characterization of hOMESC-derived hiPSCs:
The EB formation test showed that the generated hiPSC lines could differentiate in all the three germ layers. In fact, after the simultaneous removal of both adhesion stimuli and b-FGF to trigger random differentiation, hOMESC-derived hiPSCs expressed transcripts of markers belonging to the ectoderm (TUBB and PAX6), mesoderm (FLK1 and PECAM), and endoderm (AFP and GATA4) layers (Fig. 4D).
hOMESC-derived hiPSCs can be differentiated into corneal keratinocyte-like cells
To test the feasibility of using EEC-hiPSCs to generate corneal epithelium, wt- and R279H-hiPSCs were differentiated toward corneal epithelium using a modification of a previously published protocol (Mikhailova et al., 2014). As expected, at day 0 of differentiation, hiPSC lines were negative for epithelial keratins and expressed the pluripotency marker OCT4 that was lost within 1 week of differentiation (Fig. 5). Starting from week 1, differentiating cells began to stain positive for keratins and showed an evident keratinocyte-like morphology that increased with time, indicating their epithelial commitment (Fig. 5). As early as differentiating day 10, both wt- and R279H-hiPSC lines started to express the multipotent progenitor marker ΔNp63α, indicating progenitor commitment (Fig. 6A).
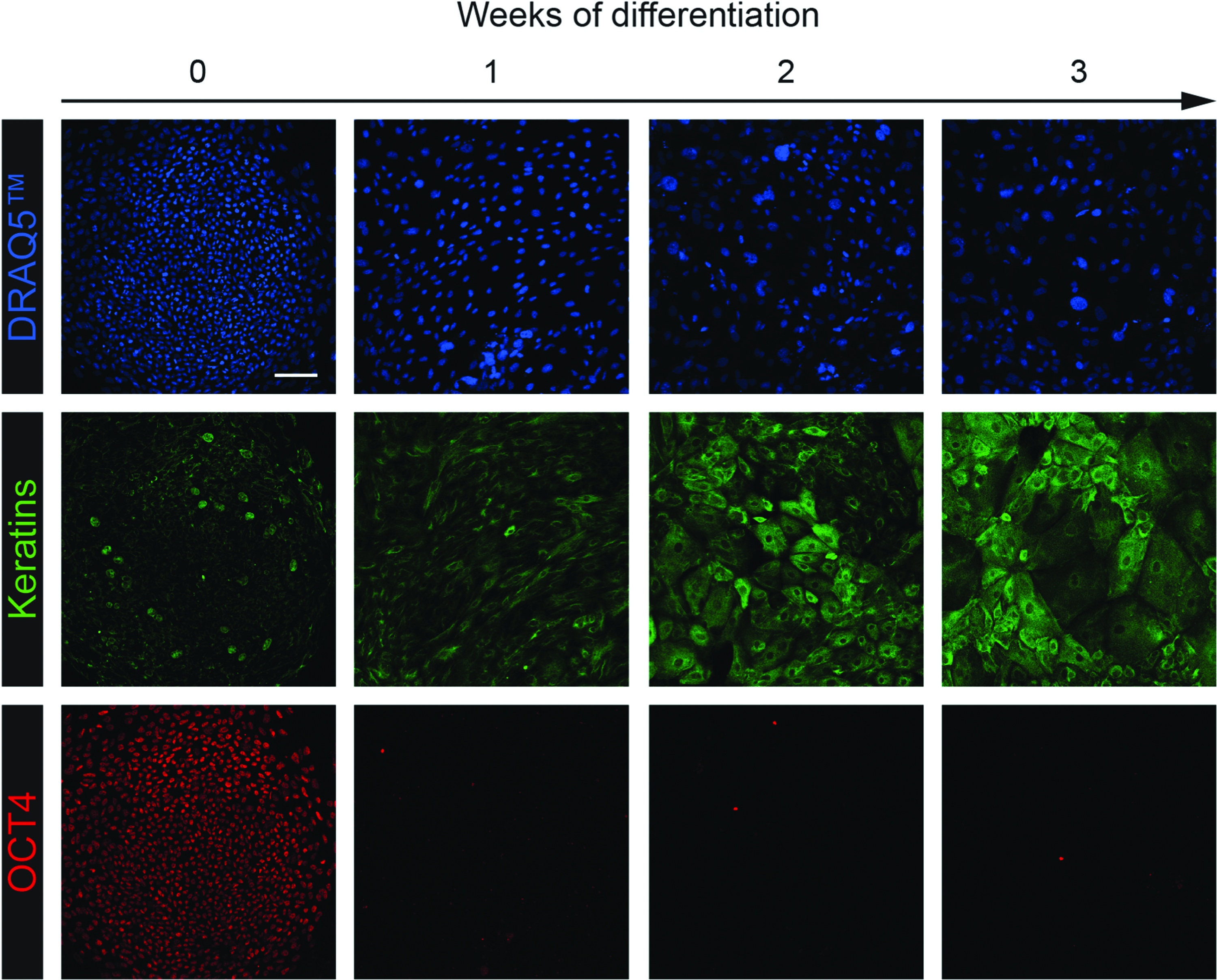
Differentiation of hOMESC-derived hiPSCs into corneal epithelium. hOMESC-derived hiPSCs were differentiated toward corneal epithelium as described in the Materials and Methods section. At different weeks of differentiation (horizontal arrow), cells were fixed and immunostained to detect nuclei (DRAQ5, top panels), keratins (middle panels), and OCT4 (bottom panels). Images were acquired by CLSM using a Nikon A1 microscope (scale bar = 100 μm). Color images available online at www.liebertpub.com/cell
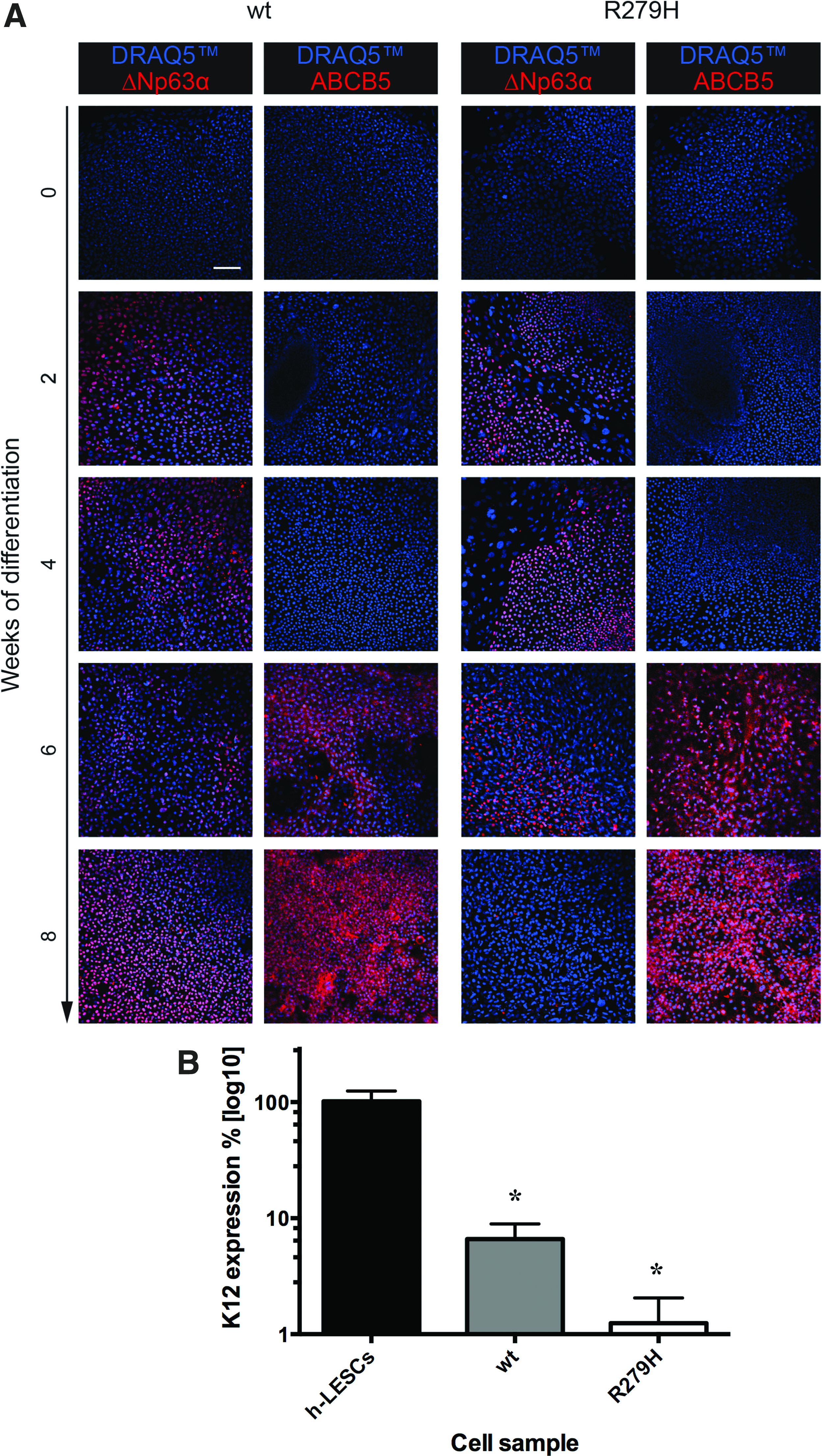
Expression of putative corneal markers by differentiating hOMESC-derived hiPSCs.
By week 6, the limbal stem cell marker ABCB5 started to be detectable (Fig. 6A). Importantly, both wt and R279H- differentiating hiPSCs started to express by day 30 corneal specific marker Keratin 12 (K12), although to lower levels compared to h-LESCs (Fig. 6B), further demonstrating their corneal commitment.
In conclusion, these results demonstrate that hiPSCs can be generated from EEC-hOMESCs by two different integration-free approaches and that the derived EEC-hiPSCs can be differentiated toward corneal epithelium underlying the potential to be used for disease modeling and for patient-specific cell and gene therapy of corneal defects.
Footnotes
Acknowledgments
This study was funded by the Italian Ministry of Health (grant No. RF-VEN-2008-1200634 to Luisa Barzon), by AFM-Association Francaise contre les Myopathies (grant No. 20687 to Enzo Di Iorio), and by University of Padova (grant No. CPDA159895/15 to Enzo Di Iorio, No. 60A07-2142/15 to Marta Trevisan, and 60A07-1024/15 to Gualtiero Alvisi).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.