
Abstract
Abstract
Establishing pig embryonic stem cells (pESCs) remains a challenge due to differences in the genetic backgrounds of mouse, human, and pig. Therefore, pig-specific pluripotency markers and cellular signaling must be identified. In this study, doxycycline (DOX)-inducible vectors carrying Oct4, sex-determining region Y-box 2 (Sox2), Nanog, Kruppel-like family 4 (Klf4), or Myc, which are known reprogramming factors, were transduced into pESCs. And pluripotency genes were analyzed in one or two reprogramming factor-expressed pESCs. When cultured without DOX, pESCs were stably maintained in basic fibroblast growth factor-supplemented media. However, when treated with DOX, the cells lost their alkaline phosphatase (AP) activity and differentiated within 2 weeks. Subsequently, we investigated the expression of genes related to pluripotency in DOX-treated pESCs using quantitative reverse transcription-polymerase chain reaction (PCR). Expression levels of Oct4, E-cadherin, and Fut4 were significantly increased by Oct4 overexpression, and Oct4 and Fut4 were upregulated in the Sox2-infected group. When a combination of two reprogramming factors, including Oct4 or Sox2, was introduced, weak AP activity remained. In addition, several of the two reprogramming factor transduction groups could be maintained after subculturing with transgene activation. Although long-term culture failed, pESCs transduced with Oct4 and Nanog, Oct4 and Klf4, or Sox2 and Nanog combinations could be subcultured even under transgene activation conditions. Analysis of the cause of long-term culture failure by quantitative PCR confirmed that the expression of intermediate reprogramming markers was not maintained. Given these results, additional methods are needed to support the completion of each reprogramming phase to succeed in the conversion of the pluripotent state of pESCs. This study improves our understanding of pluripotent networks and can be used to aid in the establishment of bona fide pig pluripotent stem cells.
Introduction
P
However, these markers were based on markers of undifferentiated mouse ESCs (mESCs), and studies on pig-specific markers are lacking. pESCs are primed pluripotent stem cells that can differentiate into all three germ layers in vitro (Piedrahita et al., 1990, 1999; Wianny et al., 1997), but fail to show chimeric complementation because the pluripotency level of pESCs is not as high as that of the naive pluripotent cells (Mueller et al., 1999; Piedrahita et al., 1998; Shim et al., 1997). There is a lack of research on the specific characteristics of pESCs.
Recently, a number of studies have reported that pluripotent stem cells can be categorized as naive or primed according to the level of pluripotency (Hanna et al., 2010; Nichols and Smith, 2009). mESCs represent naive pluripotent stem cells, whereas ESCs of human and large animal species, such as rabbits, dogs, pigs, sheep, goats, and nonhuman primates, and mouse epiblast stem cells (mEpiSCs) are considered to be in the primed state. Naive pluripotent cells form dome-shaped colonies and are self-renewing by leukemia inhibitory factor (LIF) signaling and inhibition of glycogen synthase kinase (GSK) and extracellular signal-regulated kinase (ERK) pathway. In addition, ESCs obtained from female cells have two active X chromosomes.
In contrast, primed pluripotent stem cells form flat colonies and undergo self-renewal by fibroblast growth factor (FGF) signaling. Primed pluripotent stem cells show a greater differentiated status than naive pluripotent stem cells in terms of developmental capacity, gene expression, and epigenetic signature.
Thus, naive pluripotent cells are easier to use than primed pluripotent stem cells for genetic engineering. Naive pluripotent stem cells can be injected into recipient blastocysts to produce chimeric embryos. However, primed pluripotent stem cells have failed in vivo differentiation assays through blastocyst injection. Together, these characteristics indicate that naive pluripotent stem cells have a higher degree of pluripotency than primed pluripotent stem cells. Thus, many studies have been conducted on the conversion of primed pluripotent cells into naive pluripotent cells (Bao et al., 2009; Guo et al., 2009; Yang et al., 2010).
Many recent studies have compared the conversion of primed pluripotent stem cells into naive pluripotent stem cells and the reprogramming of somatic cells into induced pluripotent stem cells (iPSCs). The reprogramming of somatic cells into iPSCs requires sequential steps. When reprogramming factors are introduced into somatic cells, Ccnd1, Ccnd2, and genes involved in DNA replication, which are involved in cell proliferation, are expressed at an early stage. This increases the ratio of the S phase in the cell cycle. The genes expressed in somatic cells (Snai1, Snai2, and Thy1) are downregulated by histone modification. Then, the reprogrammed cells in the early stage are transferred to the intermediate stage, and their shape becomes rounder and denser (Koche et al., 2011; Mikkelsen et al., 2008; Samavarchi-Tehrani et al., 2010).
Inside the cells, TGF-β signaling is suppressed and promesenchymal-to-epithelial transition (MET) microRNAs are activated. As a result, bone morphogenetic protein (BMP) signaling is enhanced and MET occurs. Cells at this stage exhibit stochastic activation of markers associated with pluripotency (Buganim et al., 2012), and developmental regulators (Polo et al., 2012) and glycolysis are activated (Hansson et al., 2012). After this step, genes such as undifferentiated embryonic cell transcription factor 1, estrogen-related receptor beta (Esrrb), developmental pluripotency associated 2, sal-like protein 4 (Sall4), and Lin28 are highly expressed, which activate sex-determining region Y-box 2 (Sox2). The activity of Sox2 causes deterministic events either directly or indirectly.
In the late phase, the cytoskeleton redesigns the cell morphology into an ESC-like shape. Repressive chromatin characteristics, such as H3k27 or H3k9, disappear from the pluripotency core transcription factors, activating pluripotency circuitry. When pluripotency circuitry is activated, Oct4, Nanog, Rex1, and Esrrb are highly expressed, and the pluripotent state is maintained even when the reprogramming factors that were introduced ectopically are removed (Buganim et al., 2012; Golipour et al., 2012; Hansson et al., 2012; Polo et al., 2012).
In this study, we overexpressed reprogramming factors in primed pESCs and analyzed the changes that occurred in endogenous genes related to pluripotency. This study improves our understanding of pluripotent networks and can be used to aid in the establishment of bona fide pig pluripotent stem cells.
Materials and Methods
Animal care
The care and experimental use of pigs and mice were approved by the Institutional Animal Care and Use Committee at Seoul National University [Approval No.: ILAR-17-06-192 for mouse embryonic fibroblast (MEF) isolation]. The ovaries used in this study were donated from local slaughterhouses (Dodram, Korea; Samsung, Korea) for research purposes. Pregnant Institute of Cancer Research (ICR) mice were purchased from Samtaco Bio, Inc., Korea. The mice were cared for according to the standard protocol of the Institute of Laboratory Animal Resources and sacrificed by cervical dislocation after anesthesia.
Lentiviral transduction of reprogramming factors
Production and transduction of lentiviral vectors with inducible systems containing human Oct4, Sox2, Klf4, Myc, and pig Nanog were performed following the protocols described by Choi et al. (2016).
Culture and growth of pESCs
pESCs were derived from blastocysts produced in vitro. The produced and hatched blastocysts were seeded onto feeder cells composed of mitotically inactivated MEFs according to previous studies (Park et al., 2013; Son et al., 2009). After 5–7 days, primary colonies of ESCs were observed and cultured for ∼7–10 days. Fully expanded colonies were mechanically dissociated by trituration with pulled-glass pipettes and transferred onto new feeder cells for subculturing.
pESCs were cultured in a pESC medium (PESM) consisting of Dulbecco's modified Eagle's medium (DMEM; low glucose), Ham's F10 medium containing 15% fetal bovine serum (FBS; collected and processed in the USA), 2 mM GlutaMAX, 0.1 mM β-mercaptoethanol, and 1 × antibiotic/antimycotic (Gibco). To support pluripotency and self-renewal, the ESCs were cultured in PESM with 20 ng/mL human recombinant basic FGF (bFGF) (R&D Systems) and 100 ng/mL heparin sodium salt (Sigma-Aldrich).
For the analysis of overexpressed genes, the transfected cells were cultured in medium supplemented with 2 ng/mL doxycycline (DOX) and 1000 U/mL LIF (Millipore, MA). The medium was changed every 24 hours and all cells were cultured in humidified conditions with 5% CO2 at 37°C. pESCs were subcultured every 5–7 days using pulled-glass pipettes. Expanded colonies were detached from the feeder cells and dissociated into small clumps. They were transferred onto new feeder cells containing mitomycin C-treated (Roche, Germany) MEFs.
AP staining
AP staining was performed using the nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate toluidine salt (NBT/BCIP; Roche) method. Before staining, all cell samples were preincubated for 10 minutes at 4°C and fixed with 4% paraformaldehyde for 20 minutes. After washing, fixed cells were incubated for 30 minutes at room temperature in the dark with NBT/BCIP stock solution diluted in a buffer solution (0.1 M Tris-HCl and 0.1 M NaCl, pH 9.5). Cells were examined under an inverted microscope.
Embryoid body formation and in vitro differentiation of pESCs
To evaluate the capacity for in vitro differentiation, embryoid bodies (EBs) were generated from pESCs. Cultured ESC colonies were detached from the feeder cells and colonies were mechanically dissociated into small clumps. These clumps were cultured in Petri dishes without cytokines for 5 days. After suspending the culture, the dissociated cells aggregated and formed EBs. The cultured EBs were seeded onto 0.1% gelatin-coated plates and further cultured for 2 weeks with DMEM containing 15% FBS. After 2 weeks, differentiated cells were analyzed using reverse transcription-polymerase chain reaction (RT-PCR), quantitative real-time PCR (qPCR), and immunostaining with differentiation-specific antibodies.
Immunocytochemistry staining
Immunocytochemistry analyses were performed to evaluate the expression of genes related to pluripotency and differentiation. Before staining, all cell samples were preincubated for 10 minutes at 4°C and fixed with 4% paraformaldehyde for 30 minutes. After washing twice with Dulbecco's phosphate-buffered saline (DPBS; Welgene), samples were treated for 1 hour with 10% goat serum in DPBS to prevent nonspecific binding. Serum-treated cells were incubated overnight at 4°C with the primary antibodies.
The primary antibodies were as follows: Oct4 (1:200; Santa Cruz Biotechnology, CA), Sox2 (1:200; Millipore), Nanog (1:200; Santa Cruz Biotechnology), Neurofilament (1:100; Millipore), Vimentin (1:100; Millipore), and Cytokeratin17 (1:100; Millipore). When using antibodies for intracellular proteins such as Oct4, Sox2, and Nanog, fixed cells were treated for 5 minutes with 0.2% Triton X-100 (Sigma-Aldrich) before serum blocking. After incubating with the primary antibody, the cells were treated for 3 hours at room temperature with Alexa Fluor-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342 (Molecular Probes). Images of the stained cells were captured using an inverted microscope.
Reverse transcription-polymerase chain reaction
Total RNA was extracted from the cells using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized using the High-Capacity RNA-to-cDNA Kit. cDNA was amplified with 2 × PCR master mix solution (iNtRON, Korea) and 2 pmol of primers, as shown in Tables 1 and 2. PCR reactions were performed in a thermocycler under the following conditions: 94°C for 5 minutes, 35 cycles of denaturation at 95°C for 30 seconds, annealing for 30 seconds (where the annealing temperatures depended on each primer set), extension at 72°C for 30 seconds, and a final extension at 72°C for 7 minutes. Amplified PCR products were visualized using electrophoresis on 1% agarose gel stained with ethidium bromide.
Quantitative real-time polymerase chain reaction
To verify the gene expression levels in pESCs, we performed qPCR. Total RNA from individual samples was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized using the High-capacity RNA-to-cDNA Kit (Applied Biosystems, CA) according to the manufacturer's instructions, and a final volume of 20 μL of cDNA was produced. Extracted cDNA samples were amplified with the DyNAmo HS SYBR Green qPCR Kit (Thermo Scientific, MA) containing 1 pmol of each primer set listed in Table 3 in a 10-μL reaction volume.
Amplification and detection were conducted using the ABI 7300 Real-Time PCR System (Applied Biosystems) under the following conditions: one cycle of denaturation at 50°C for 2 minutes and 95°C for 10 minutes, 40 cycles of denaturation at 95°C for 15 seconds, and annealing/extension for 1 minute (where the annealing/extension temperatures depended on each primer set). The relative expression level was calculated by normalizing the threshold cycle (Ct) values of each gene to that of ACTB using the Δ−Ct method (Livak and Schmittgen, 2001).
Statistical analysis
The gene expression data from the qPCR analysis data were statistically analyzed using GraphPad Prism 6 statistical software (GraphPad Software, CA). All statistical differences in the datasets (except those presented in Fig. 2E) were determined using one-way analyses of variance (ANOVAs) followed by Fisher's least significant difference test. The statistical differences in the data presented in Figure 2E were investigated using ANOVA followed by the Bonferroni post hoc test. Differences were considered significant when p < 0.05.

Reprogramming factor-transduced pESC morphology and AP activity.
Results
Characterization of pESCs
The pESC line used in this study was generated from porcine preimplantation blastocysts using in vitro fertilization in a manner reported previously (Lee et al., 2007). The established pESC line was flat, similar to previous reports of human ESCs (hESCs) and mouse EpiSCs (Fig. 1A) (Park et al., 2013). In addition, they possessed AP activity (Fig. 1B) and were stably maintained in their pluripotent state over long periods (>40 passages in 1 year). The cells were analyzed for pluripotent marker expression and their capacity for in vitro differentiation according to previously reported standards (Park et al., 2013). Expression of transcription factors related to pluripotency such as Oct4, Sox2, and Nanog was detected using immunocytochemistry (Fig. 1C).
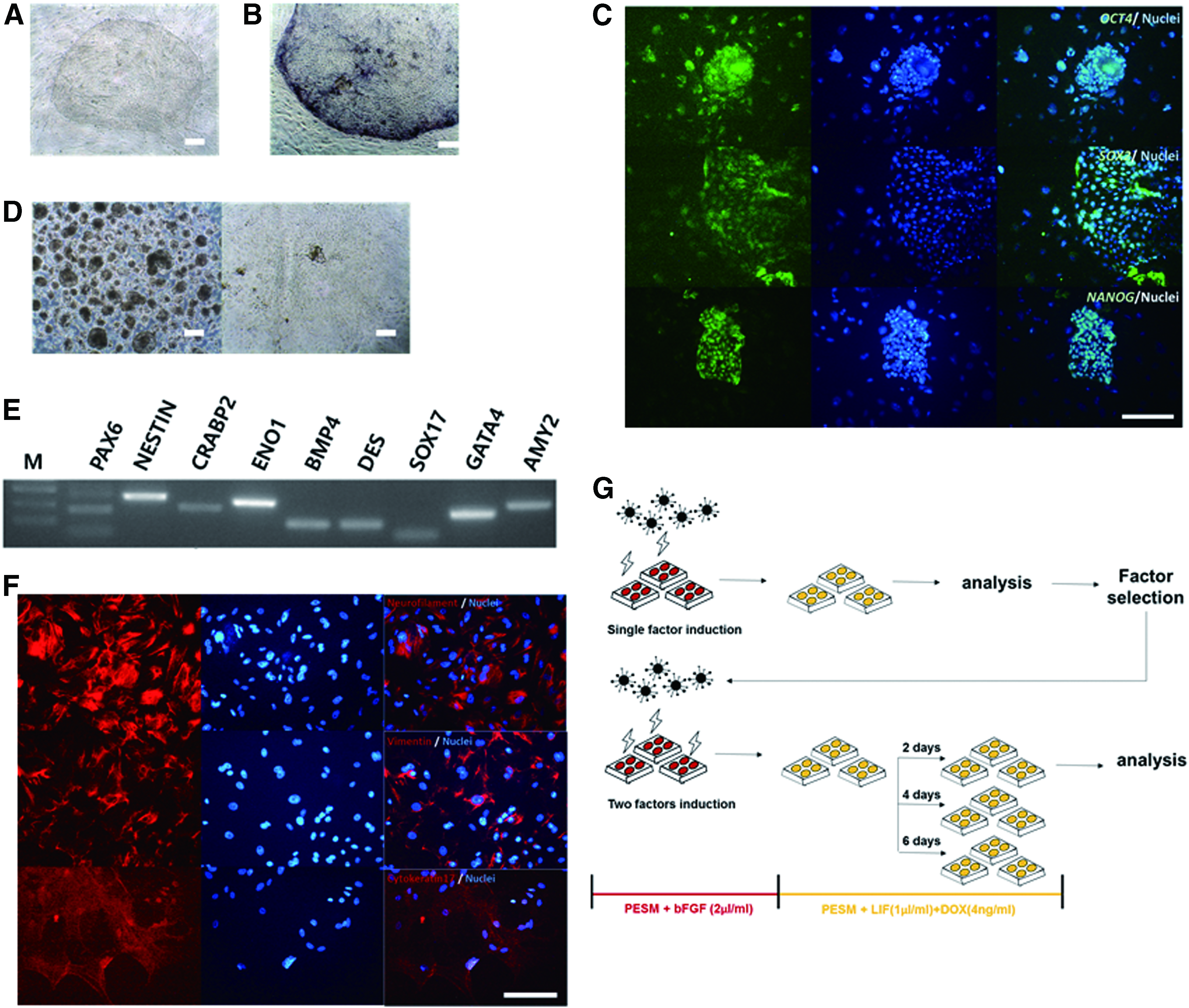
Characterization of pESCs.
The cells were detached from feeder cells and cultured in suspension to verify their capacity for differentiation. The pESCs cultured in suspension formed EBs (Fig. 1D, left panel). The generated EBs spontaneously differentiated into the three germ layers upon placement on gelatin-coated plates (Fig. 1D, right panel). In the differentiated cells, the expression of differentiation marker genes in the three germ layers was detected by RT-PCR and immunostaining (ectoderm: PAX6, NESTIN, CRABP2, and neurofilament; mesoderm: ENO1, BMP4, DES, and vimentin; and endoderm: SOX17, GATA4, AMY2, and cytokeratin17). Thus, we confirmed that the established cell line was pluripotent with the differentiation potential to generate the three germ layers (Fig. 1E, F).
We performed an experiment to observe the changes that occurred when reprogramming factors were overexpressed in the pESCs. First, DOX-inducible vectors carrying Oct4, Sox2, Nanog, Klf4, or Myc, known as reprogramming factors, were transduced into pESCs. The cells were stabilized and then cultured in a medium supplemented with LIF and DOX or bFGF and DOX.
Some of the single-gene overexpression groups were used for gene expression analysis and others were maintained for morphological observations. Based on the results of the gene expression analysis, combinations of two reprogramming factors were introduced into the pESCs and the culture medium was replaced with LIF- and DOX-supplemented medium. The two-gene transduced pESCs were cultured in an exchanged medium for 2, 4, or 6 days, and then sampled for analysis of endogenous gene expression patterns and pESC morphology (Fig. 1G).
Observation of pESCs with single-factor overexpression
Initially, we hypothesized that pESCs have lower pluripotency or different pluripotency mechanisms compared to hESCs. Therefore, to verify a possibility of pluripotent state conversions, we examined changes of endogenous pluripotency genes in all four Yamanaka factor-introduced pESCs (Supplementary Fig. S1A, B; Supplementary Data are available online at www.liebertpub.com/cell). Experimental results showed that expression of reprogramming markers increased as reprogramming progressed, and naive status markers were detected by immunostaining in Yamanaka's factor-overexpressed pESCs. As Yamanaka's factors affected pluripotency genes in pESCs, we tried to find roles of reprogramming factors and convert pluripotent state in pig stem cells. A single reprogramming factor was introduced into pESCs using the tetracycline-inducible system (Fig. 2D).
In the Tet-Off state, the pESCs with single-factor overexpression formed flat colonies and exhibited AP activity, similar to typical pESCs. However, when DOX was added to the medium to activate the transgene, the pESCs lost their typical characteristics. The cell mass of the pESCs decreased gradually, the boundary of the colony became obscured, and AP activity was lost (Fig. 2B). As the subculture progressed, the transgene-activated experimental groups became differentiated (Fig. 2C).
To analyze these results based on endogenous gene expression, we performed qPCR (Fig. 3A–H). Before this experiment, we found that lentiviral infection had no effect on gene expression (Fig. 2E). The pESCs with single-factor overexpression were cultured for 3 days on LIF- and DOX-supplemented medium, because long-term culture was not possible. We selected gene candidates for qPCR based on a reprogramming process (early phase: E-cadherin; intermediate phase: Lin28 and Sall4; and late phase: Rex1, Fut4, Klf2, and Dax1) and a representative pluripotent marker (Oct4) (Buganim et al., 2012; Golipour et al., 2012; Hansson et al., 2012; Polo et al., 2012). Analysis of the qPCR results revealed that most ectopic factors did not downregulate endogenous genes in the pESCs.
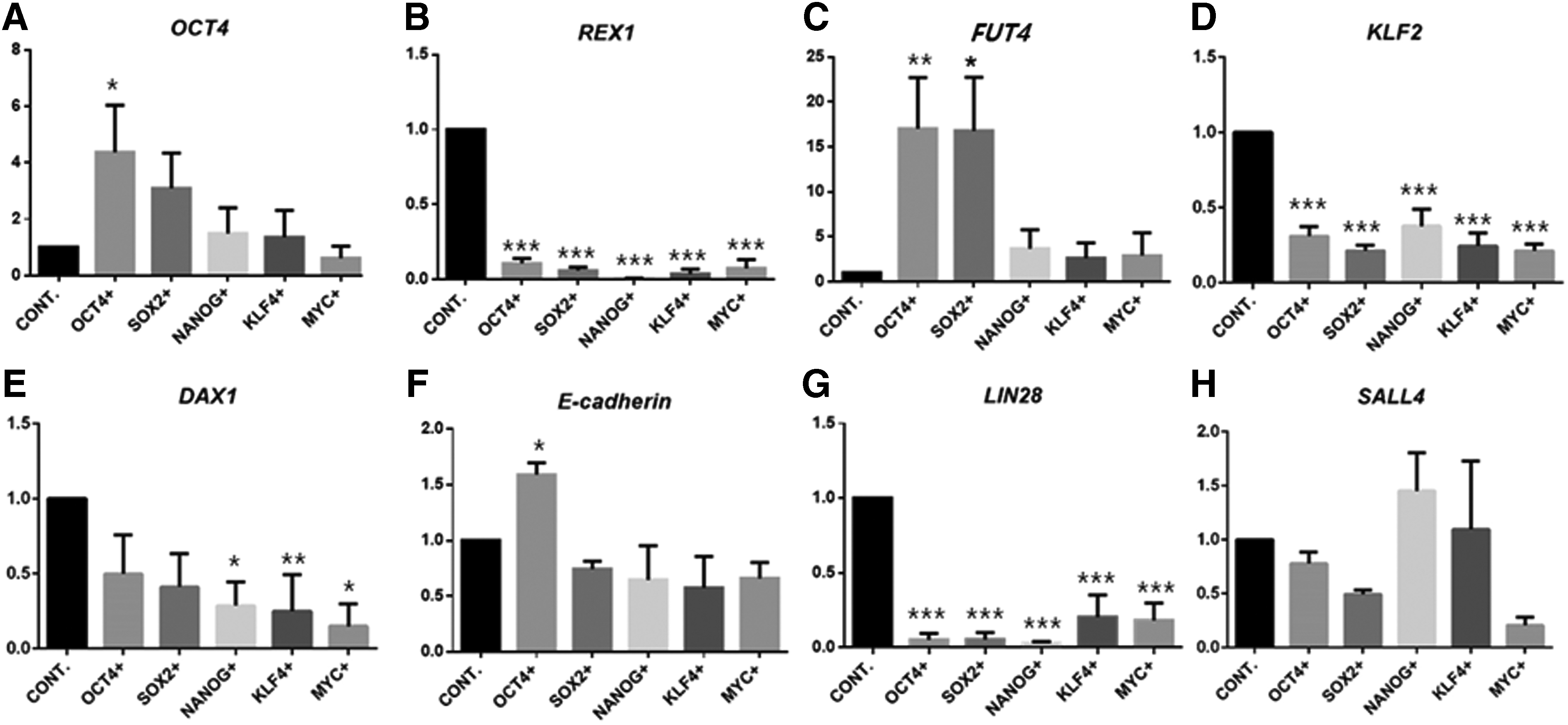
Analysis of pluripotent-related gene expression using qRT-PCR. Three days after treatment with LIF and DOX, the expression of endogenous pluripotent-related genes, including
However, the experimental group with Oct4 and Sox2 exhibited significant upregulation of several endogenous genes in pESCs. The Oct4 overexpression group induced upregulation in endo-Oct4, Fut4, and E-cadherin, and the Sox2 overexpression group significantly upregulated endo-Fut4.
Analysis of pESCs with two overexpressed factors
Based on the analysis of overexpressed pESCs, we transduced two factors into the pESCs. DOX-inducible lentiviral vectors with a combination of Oct4 and Sox2, Oct4 and Nanog, Oct4 and Klf4, or Sox2 and Nanog were introduced into pESCs (Supplementary Fig. S1C). Typical colony morphology and AP activity of pESCs were observed in the Tet-Off state (Fig. 4A).

Morphology and AP activity of two reprogramming-factor-transduced pESCs.
However, when the transgene was activated using LIF- and DOX-supplemented media, the pESCs with two overexpressed factors showed a different appearance from the control group, including decreased cell mass and AP activity (Fig. 4B). Interestingly, when one reprogramming factor was introduced, the AP activity was lost completely, but when two reprogramming factors were introduced, a weak AP activity remained. In addition, several of the groups transduced with two reprogramming factors could be maintained after subculturing, with transgene activation.
Although long-term culture failed, pESCs with Oct4 and Nanog, Oct4 and Klf4, or Sox2 and Nanog could be subcultured even under transgene activation conditions. We were able to observe the colonies for about 2 weeks. However, the remaining groups were differentiated after subcultivation in transgene-activated states (Fig. 4C). Based on these results, we observed changes in endogenous genes related to pluripotency when two reprogramming factors were overexpressed in pESCs. Transgenes were activated and qPCR was performed in samples collected after culturing for 2, 4, and 6 days. In total, four groups, including the three experimental groups that could be subcultivated in the transgene activation state and a group with Oct4 and Sox2, were observed.
The analysis of the qPCR results showed that E-cadherin, an early-phase reprogramming marker, was upregulated on day 2, and then decreased slightly on day 4 and maintained a similar level on day 6. Conversely, the intermediate-phase reprogramming markers Lin28 and Sall4 increased more than 10-fold on day 2, and 40-fold in the Sox2 and Nanog transduction group. However, on day 4, they tended to decrease sharply and showed little expression on day 6. The late-phase reprogramming markers Fut4, Rex1, and Klf2 were not expressed throughout the experiment. Meanwhile, Dax1 was upregulated on day 2, but showed little expression on days 4 and 6 (Fig. 5A–H).
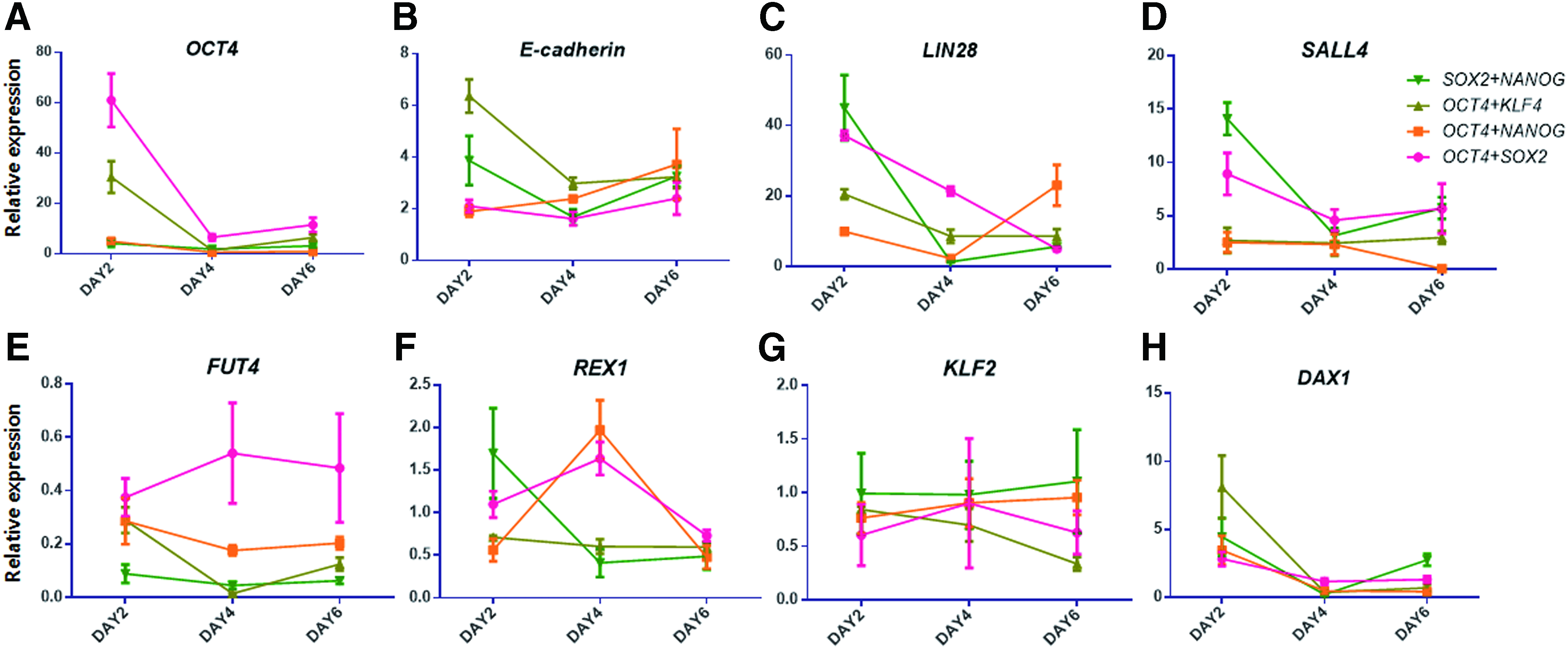
qRT-PCR analysis of pluripotent-related genes in two-factor-transduced pESCs. To verify the effects of ectopic gene expression on endogenous genes, expression of endogenous genes representing pluripotency and the reprogramming phase [pluripotency marker: Oct4
Discussion
Expression of genes related to pluripotency could not be maintained in pESCs with overexpressed reprogramming factors
The experimental groups overexpressing one reprogramming factor in pESCs may have differentiated because of an imbalance in their pluripotency network. Pluripotency networks are balanced by a number of genes with complex mechanisms (e.g., antagonism, synergistic action, and additive interaction). Niwa et al. (2000) reported that Oct-3/4 expression should be maintained at an appropriate level for mESCs to remain undifferentiated. In this study, a twofold increase in the expression of Oct-3/4 in mESCs led to differentiation into primitive endoderm and mesoderm. Conversely, after (Niwa et al., 2000) lowering the expression Oct-3/4 level, the cells differentiated into trophectoderm (TE).
In another study, Nanog was overexpressed in mEpiSCs and hESCs. The results of this study suggested that hESCs with overexpressed Nanog failed to differentiate into ectoderm and definitive endoderm. Based on the results of this experiment and previous studies, if the precise expression levels of reprogramming factors in pESCs are not maintained, the pluripotent balance can be broken. Meanwhile, mEpiSCs transduced with Klf4 resulted in the successful formation of germline-competent chimeras and naive marker expression (Guo et al., 2009). In addition, Klf2 was introduced into mEpiSCs instead of Klf4 and cultured with GSK and ERK pathway inhibitor (2i) and LIF to support the conversion into a naive-like state (Hall et al., 2009a).
Together, these studies show that even in the same primed pluripotent state, cells differ in their pluripotent networks depending on species and strains.
pESC conversion occurred in the intermediate stage, but was not completed
Although pESCs with one overexpressed reprogramming factor were not maintained in this experiment, the results showed that Oct4 and Sox2 acted as triggers for the transient high expression of endogenous Oct4, Fut4, and E-cadherin in pESCs. In many studies, Oct4 has been shown to have an important role in the maintenance of pluripotency and reprogramming of differentiated cells (Li et al., 2010; Vallier et al., 2009; Zafarana et al., 2009). Oct4-centered protein interaction analysis results and the genome-wide identification results of the Oct4 target gene indicate that Oct4 co-binds with pluripotency regulators. These results demonstrate that Oct4 has an important role in maintaining the undifferentiated state of pluripotent cells (Pardo et al., 2010; van den Berg et al., 2010).
Sox2 is another essential element in maintaining the pluripotent cells, as verified by Sox2 null mESCs that differentiated into TE-like cells because Sox2 is related in the maintenance of orphan nuclear receptors such as Nr5α2. Nr5α2 is associated with the activity of Oct4 combined with the Oct4 proximal promoter (Masui et al., 2007). In addition, endogenous Sox2 expression is an event necessary to reprogram somatic cells into miPSCs. Endogenous Sox2 expression induces the reprogramming of the next event to occur and the expression of genes related to pluripotency such as Lin28, Esrrb, and Sall4 (Buganim et al., 2012). These results and previous reports suggest that Oct4 and Sox2 are key transcription factors in pluripotency networks, and that they interact with a variety of genes related to pluripotency.
Based on the experimental results shown in Figure 3, all of the two-factor transduction pESC treatment groups failed long-term culturing. The qPCR analysis (Fig. 5) offered several reasons for this failure. Self-renewal by LIF signaling is a characteristic of cells with naive pluripotency. Therefore, to cultivate primed pESCs with primed pluripotency in LIF-supplemented media, reprogramming is required. However, in the pESCs transduced with two reprogramming factors, the expression of genes expressed at the intermediate phase of reprogramming was not maintained. E-cadherin, which was expressed early in reprogramming, showed an mRNA expression level increase by sixfold or more on day 2, which decreased slightly on day 4 and was maintained until day 6.
However, Lin28 and Sall4, which are expressed in the intermediate phase of reprogramming, were expressed only on day 2, and then decreased rapidly and were not expressed on day 6. Therefore, the naive marker genes Rex1, Fut4, and Klf2 were not expressed after day 2. These results demonstrate that reprogramming factor transduction in pESCs failed to induce reprogramming.
Converting the pluripotency state of pESCs requires gene overexpression combined with additional methods
Like pESCs, the pluripotency states of hESCs and mEpiSCs with primed pluripotency have been successfully converted by introducing two reprogramming factors (Hanna et al., 2010; Silva et al., 2009; Yang et al., 2010). However, the reprogramming conditions of mouse and humans cells do not apply to pigs, because the molecular mechanisms during embryo development differ. Pigs require a longer period of time before embryos are implanted than mice and humans (Alberio and Perez, 2012).
Therefore, the signaling that regulates the pluripotency of pig inner cell mass (ICM) differs from that of mouse embryo. Unlike mouse, pigs express Oct4 and Cdx2 in ICM and TE. Conversely, Sox2 is expressed from the beginning of embryo cavitation, and when it is divided into TE/ICM, it appears specifically only in the embryonic portion and gradually disappears with the progression of differentiation. ICM cells and epiblasts of pig blastocysts do not have LIF receptors, whereas FGF receptors are highly expressed. Therefore, FGF signaling has a more important role in maintaining pluripotency in pigs than LIF signaling (Hall et al., 2009b).
In addition, strong expression of BMPr and SMAD in porcine ICM cells and EpiSCs was detected by immunostaining and BMP signaling inhibitor experiments in vitro. When pig ICM cells and EpiSCs were treated with BMP inhibitors in vitro, the survival rate of the cells decreased (Hall and Hyttel, 2014). BMP signaling also has an important role in maintaining pig pluripotency. In addition, pig embryos could form chimeric embryos by aggregated with primed hiPSCs, but not with naive rodent iPSCs (Wu et al., 2017). This result showed the state of pluripotent cells that resided in pig embryos is close to primed state rather than naive state in terms of developmental competences.
Recently, Zhang et al. (2015) succeeded in generating “intermediate” pig pluripotent stem cells with both naive and primed characteristics even when the transgene was removed using three growth factors (LIF, FGF2, and BMP4) and the inhibitor 2i. These results show that pig pluripotent stem cells maintain pluripotency by unique cell signaling that differs from those of mouse and human. The theory that divides pluripotent stem cells into two states according to their pluripotent state was first developed in mouse stem cells. Therefore, it is hard to divide the stem cells of pigs and other mammals based on the criteria.
There are cases of the successful establishment of pig naive-like pluripotent stem cells by introducing two reprogramming factors. Telugu et al. (2011) transduced Oct4 and Klf4 into the ICM of porcine blastocysts and isolated pESCs. These pESCs were self-renewed by LIF, and their ability to differentiate into the three germ layers was verified using the teratoma formation assay (Telugu et al., 2011). This suggests that transducing reprogramming factors to establish porcine naive-like pluripotency cells by reprogramming factor transduction methods can be effectively used to gain a pluripotent cell source with low epigenetic memory.
Recently, the results of producing chimeric offspring by using six human reprogramming factor (Oct4, Sox2, Nanog, Klf4, Lin28, and c-Myc)-transduced pig iPSCs were also reported (West et al., 2010). Overall, these results indicate that pig-specific culture systems or reprogramming methods are required to obtain pig naive pluripotent stem cells.
Conclusions
In this study, all of the experimental groups in which one or two reprogramming factors were transduced into pESCs failed to show long-term cultivation in an LIF-supplemented medium. Based on qPCR analysis performed to determine the reasons for this failure, the expression of both Lin28 and Sall4 genes was not maintained. Since these two genes are markers expressed at the intermediate stage of reprogramming, it can be presumed that the expression of both genes was not maintained because the intermediate stage of reprogramming was not completed.
Based on these results, additional methods are required to complete each reprogramming phase to succeed in converting pESCs into a pluripotent state. Overall, to support the conversion of pESCs into a pluripotent state, a greater understanding of the process is required to identify the unique pluripotency network in pig and determine whether bona fide pig pluripotent stem cells can be established.
Footnotes
Acknowledgment
This work was supported by BK21 Plus Program and the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2017R1D1A1B03032256).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.