
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) play an important role in cell replacement therapy. Several studies have shown that keratinocytes are promising reprogrammed cells. We easily and efficiently enriched epidermal stem cells by attaching them for a limited time in culture dishes. Individual epidermal cells enriched in stem cells, which showed strong immunostaining for K15, were obtained and generated iPSCs within 10 days after transfection with lentiviruses encoding 4 transcription factors (OCT4, SOX2, KLF4, and NANOG). Immunofluorescent staining showed that those iPSCs expressed SOX2, OCT4, NANOG, and SSEA3 (a specific marker of embryonic stem cells). The embryoid bodies generated from those iPSCs stained positively for OCT4 and NANOG and also with the CDy1 dye that is specific for stem cells. When the iPSCs were subcutaneously injected into 4-week-old BALB/c nude mice, teratoma developed at the inoculation site. The iPSCs also demonstrated reduced DNA methylation compared with the original cells and could be induced to differentiate into adipocytes (mesodermal), hepatocytes (endodermal), and neural cells (ectodermal) in vitro. Our research provides an easy and efficient method for producing iPSCs from keratinocytes, which has important applications in cell replacement therapy.
Introduction
A
For therapeutic purposes, the cell type chosen for iPSC technology has to be easily accessible and an appropriate number of cells is needed to start efficient reprogramming. The skin is the largest organ of the body and its cells, including dermal fibroblasts, keratinocytes, dermal papilla cells, and melanocytes, can be easily isolated by using a small skin biopsy or plucked hairs (Gentile et al., 2017; Guo et al., 2011). Keratinocytes account for most of the cells in the epidermis and provide the greatest number of easily obtainable proliferating cells. Keratinocytes have emerged as a promising cell source for reprogramming, not only because of their easy availability but also due to their higher reprogramming efficiency (Raab et al., 2014). The reprogrammed percentage of fibroblasts is about 0.01%–0.5%, and the entire process requires approximately 3–5 weeks (Fidan et al., 2016).
In contrast, keratinocyte-derived iPSC colonies reach an appropriate size for passaging within 2–3 weeks, with a 100-fold (about 1%–2%) greater efficiency compared with fibroblasts when used in the various iPSC protocols (Aasen et al., 2008). The higher reprogramming efficiency of keratinocytes is mainly due to their higher basal expression levels of KLF4 and c-MYC (Khazaei et al., 2016).
Previous studies have indicated that progenitor cells show a higher reprogramming efficiency, possibly due to the fewer stochastic steps required (Yunusova et al., 2017). Human epidermis consists mainly of stratifying epithelial keratinocytes, which undergo continuous and rapid proliferation from the basal layer to the stratum corneum (Oda et al., 2018). Therefore, the inter-follicular epidermis must have a population of resident stem cells that are hidden in the basal layer that serves as a source for the steady-state cell replacement of keratinocytes. According to an improved method reported by Yang et al. (2013), we attempted to isolate putative epidermal stem cells from the skin and culture them for subsequent reprogramming. The efficiency and timeframe to form colonies were evaluated, and we further characterized the pluripotency of those iPSCs.
Materials and Methods
Isolation and culture of human epidermal keratinocytes
This study was approved by the Ethics Review Board of the Third Affiliated Hospital of Soochow University. Normal foreskin tissue from the circumcision of a 12-year-old male, taken with prior consent of the patient, was immediately immersed in phosphate-buffered saline (PBS) supplemented with 200 IU/mL penicillin-streptomycin (Cat. No. SV30010; HyClone). Under sterile conditions, after removal of subcutaneous tissue and thorough washing, the remaining tissue was trimmed into 0.5 × 0.5 cm pieces, which were then transferred to a sterile container containing 0.25% dispase (Cat. No. 4942078001; Roche) and incubated at 37°C for 2 hours.
The epidermis obtained was treated with 0.25% trypsin-EDTA (Cat. No. 25200-056; Gibco) for 15 minutes to produce a suspension of individual cells, which was passed through a 200-μm filter and then centrifuged at 1000 rpm for 5 minutes. The cell pellets were resuspended in serum-free low-calcium Epilife medium (Cat. No. EPI500CA; Invitrogen) supplemented with EpiLife Human Keratinocyte Growth Supplement (HKGS; Cat. No. S-001-5; Life Technologies), and they were then seeded in six-well culture plates.
When the cells reached 70%–80% confluence, the primary keratinocytes were detached using trypsin/EDTA (Cat. No. 25200-056; Gibco) and seeded in six-well plates coated with type IV collagen (Cat. No. C8374; Sigma) at a density of 3 × 106 cells/mL. A cover slip was placed into one well of each plate for immunohistochemistry. After 15 minutes of incubation at 37°C, some cells had adhered to the wells, and cells remaining in suspension were collected for other uses. Fresh Epilife medium was added to the adherent cells, which were further cultured at 37°C with 5% CO2. Half of the medium was replaced every 2–3 days until the cells reached 70%–80% confluence, at which time they were subcultured.
Immunocytochemistry of primary keratinocytes with K15
On the second day of subculture, the cover slip was removed from each of the 6 six-well plates and was transferred to a clean petri dish, after which they were fixed in ice cold ethanol for 10 minutes at −20°C. Immunocytochemical staining was performed using the streptavidin/peroxidase method (SP kit; Zhongshan Biotechnological Ltd. Co.). The mouse monoclonal antibody K15 (Cat. No. ab52816; Abcam) was used at a 1:100 dilution and incubated for 60 minutes at 37°C, with anti-mouse immunoglobulin G used as a secondary antibody (from the SP kit). Subsequent steps were performed according to conventional immunostaining methods. An Olympus microscope was used to observe and photograph cells.
Reprogramming of human epidermal keratinocytes
When the keratinocytes in subculture reached 70%–80% confluence, they were detached using accutase (Cat. No. A1110501;Gibco) and then transferred to a flask for further culture in fresh serum-free low-calcium Epilife medium. After 48 hours of further cultivation, a mixture of pMXs-based lentiviruses (Cat. No. 0508-107; Sidansai) encoding four transcription factors (Lenti-c-MYC-GFP, Lenti-SOX2-GFP, Lenti-OCT4-GFP, and Lenti-KLF4-GFP [1:1:1:1]) containing 10 mg/mL polybrene (Cat. No. 0832-050; Sidansai) was added to the supernatant. The flask was shaken gently on a shaker for 45 minutes at 32°C to increase the infectivity. The retroviral supernatant was removed and washed with PBS twice before replacing with fresh EpiLife medium after the shaking to prevent differentiation of the cells.
After 24 hours, the infection process just described was repeated. Two days after the second infection, the keratinocytes were trypsinized and seeded in a 12-well culture plate, which was treated with gelatin (Cat. No. G9391; Sigma) and pre-seeded with irradiated mouse embryo fibroblasts (MEFs; Cat. No. 0303-100; Sidansai) in the human iPSC medium, namely knockout (KO) Dulbecco's modified Eagle's medium (Knockout™ DMEM; Cat. No. 10829; Gibco) supplemented with 20% KO serum replacement (Cat. No. 10828; Life Technologies), 1% GlutaMAX-I (Cat. No. 25030081; Life Technologies), 1% nonessential amino acids (Cat. No. 11140; Gibco), 100 mM β-mercaptoethanol (Cat. No. 31350-100; Gibco), 1% penicillin-streptomycin, and basic fibroblast growth factor (bFGF; 10 ng/mL; Cat. No. 13256-029; Invitrogen).
After 48 hours, the medium was changed daily until iPSC colonies appeared. When the colonies grew sufficiently large, they were manually picked up with a pipette to evaluate their characteristics and were transferred into separate dishes with fresh feeder layers in iPSC medium.
Immunofluorescence and alkaline phosphatase analysis
The first-generation iPSC colonies were transferred to six-well culture plates containing cover slides and were cultured for 5–7 days. The adherent colonies were gently washed at least three times (5 minutes each) with PBS and were then fixed with 4% paraformaldehyde. Direct alkaline phosphatase (AP) activity was analyzed using a Leukocyte AP kit (Cat. No. 86R; Sigma) according to the manufacturer's guidelines.
For immunofluorescence staining of pluripotent markers, the fixed iPSC colony cells were successively incubated in 0.1% Triton X-100 (Cat. No. 9002-93-1; Sigma) in PBS for 10 minutes and then in a blocking solution containing 5% bovine serum albumin (BSA; Cat. No. 9048-46-8; Sigma) for 60 minutes at room temperature. They were then stained with the following primary antibodies that were specific for OCT4 (1:200; Cat. No. ab19857; Abcam), SOX2 (1:200; Cat. No. ab59776; Abcam), NANOG (1:100; Cat. No. ab21624; Abcam), and SSEA-3 (1:100; Cat. No. MAB4303; Millipore). All primary and secondary antibodies used were diluted in 0.1% BSA in PBS. Counterstaining with DAPI (1:1,000; Cat. No. D9542; Sigma) was performed to identify nuclei. Images were taken by using a fluorescence microscope (Olympus, Tokyo, Japan).
Analysis of DNA methylation levels
According to the manufacturer's recommendations of the Qiagen QIAamp DNA Blood Mini kit (Qiagen), genomic DNA samples were extracted from the first-generation iPSC colonies and their parental keratinocytes. An ultraviolet (UV) spectrophotometer was employed to measure the absorbance values of nucleic acids at A260 and A280. Samples with a purity of 1.6–2.1 DNA passed the bisulfate conversion quality control and were subjected to polymerase chain reaction (PCR) analysis by using primer sets designed to amplify the OCT4 and NANOG promoter regions. The template for PCR amplification was the modified DNA by bisulfate conversion. The PCR products were subcloned and then sequenced by the Shanghai Biological Engineering Co., Ltd..
Transmission electron microscopy
The first-generation iPSC colonies were manually collected, fixed in 2.5% glutaraldehyde (pH 7.2) for 4 hours, and finally post-fixed in 1% (wt/vol) osmium tetroxide for 1 hours. The specimens were dehydrated sequentially using increasing concentrations of ethanol (30%, 50%, 70%, 80%, 90%, and 100%), and they were kept in each phase for 10 minutes. The dehydrated specimens were embedded in Epon-Araldite, cut into ultrathin sections (70 nm thick) using a Leica ultramicrotome (EM UC6; Leica), mounted on copper grids, and stained with lead and uranyl solutions. Finally, the cells were observed and photographed using a JEM-2100 transmission electron microscope (TEM; JEOL, Tokyo, Japan).
Embryoid body formation
A six-well plate was coated with poly (2-hydroxyethyl)methacrylate (poly-HEMA; Cat. No. P3932; Sigma) and air-dried on a clean bench overnight. The first iPSC generation colonies were manually picked up, dispersed by accutase to individual cells, and finally cultured in suspension in a poly-HEMA-coated plate at 37°C in 5% CO2. The medium was composed of MC Medium (Cat. No. 04100; StemCell Technologies) and human iPSC medium (1:1). A volume of culture medium equal to 25% of the initial volume (0.8–1 mL) was added to the plate every day. The size and status of embryoid bodies (EBs) was observed daily using an inverted microscope, until they were suitable for further experiments, which required approximately 5–7 days. Further, a few EBs were used as adherent cultures, which were stained 1 week later with antibodies against OCT4 and NANOG, as well as the stem cell marker CDy1 dye (Active Motif).
Teratoma formation
Four-week-old BALB/c nude mice were purchased from Changzhou Cavens Laboratory Animal Co., Ltd. and were housed in specific aseptic conditions. The colonies of iPSCs were collected, washed with PBS, and digested with 0.25% trypsin/EDTA to prepare single-cell suspensions, which were subsequently centrifuged at 1500 rpm for 5 minutes. The cell pellets obtained were resuspended in a mixture of DMEM/F-12 (Cat. No. 12660-012; Life Technologies) and Matrigel (Cat. No. 354277; Biocoat) at a density of 5 × 106 cells/mL. The suspension of iPSCs (∼30 μL) was slowly injected into the subcutaneous tissue of the right rear leg of each mouse.
When the tumors grew to an appropriate size, the mice were sacrificed and the tumors were excised under sterile conditions for histopathological examination. Parts of the teratoma tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (HE).
The other parts of the teratoma were frozen and sectioned for immunohistochemical study using the following antibodies: Vimentin (1:200; Cat. No. 140417178C; Maxin), KI-67 (1:200; Cat. No. 1408041291; Maxin), OCT3/4 (1:50; Cat. No. sc-5279; Santa Cruz Biotechnology), CK(pan) (1:200; Cat. No. 140623049E; Maxin), GFAP (1:200; Cat. No. 140623049E; Maxin), and AFP (1:200; Cat. No. 13189A11; Maxin). In a sterile environment, another part of the tumor tissues was cut into pieces, digested with trypsin to obtain a single-cell suspension, routinely cultured for 7 days, and finally subjected to immunohistochemical staining with the antibodies just described.
In vitro differentiation
The first-generation iPSC clones were manually picked up and trypsinized into single-cell suspensions, which were further centrifuged and then resuspended in different differentiation media. A 24-well culture flask (Cat. No. 3473a; Corning) was pre-coated with 0.1% gelatin. The cell concentration used in these cultures was 104 cells/cm2. DMEM supplemented with 20% fetal bovine serum (Cat. No. 10099141; Gibco), 2 mM
For mesoderm differentiation, the iPSCs were cultured in differentiation medium according to the manufacturer's instructions of the Human Mesenchymal Stem Cell Functional Identification Kit (Cat. No. SC006; R&D Systems) for adipose cells. The differentiation medium contained hydrocortisone, isobutylmethylxanthine, and indomethacin. For neural induction, the EBs formed were transferred onto Matrigel-coated plates and cultured in Neurobasal Medium (1 × ; Cat. No. 21103-049; Gibco) supplemented with 1 × N2 Supplement (Cat. No. 17502-048; Life Technologies), 1 × B27 Supplement (Cat. No. 0080085-SA; Life Technologies), 1 × Insulin-Transferrin-Selenium-G (Cat. No. 41400-045; Life Technologies), Stable Glutamine (PAA), and 20 ng/mL bFGF and 20 ng/mL EGF (PeproTech) for 15 days with medium replacement every day.
On day 15, the cells were transferred onto Fibronectin-coated plates (R&D Systems) for further differentiation. Immunofluorescence staining with AFP (1:100; Cat. No. ab133617; Abcam), Oil Red O (Cat. No. 08010; Sigma), and Tubulin β-III (1:100; Cat. No. ab52623; Abcam) confirmed the induction of hepatocytes (endodermal), adipocytes (mesodermal), and neuronal cells (ectodermal), respectively.
Results
Culture of human epidermal keratinocytes
The keratinocytes derived from a young male's foreskin were cultured in serum-free and low-calcium Epilife medium, which promotes a highly proliferative and undifferentiated state. At day 14, the primary keratinocytes were approximately 70%–80% confluent and covered the bottom of the plate, with a pebble-like appearance (Fig. 1A). These cells were harvested with accutase and seeded into Type IV collagen-coated plates, and within 15 minutes, some of them had rapidly adhered to the bottom of the plate.
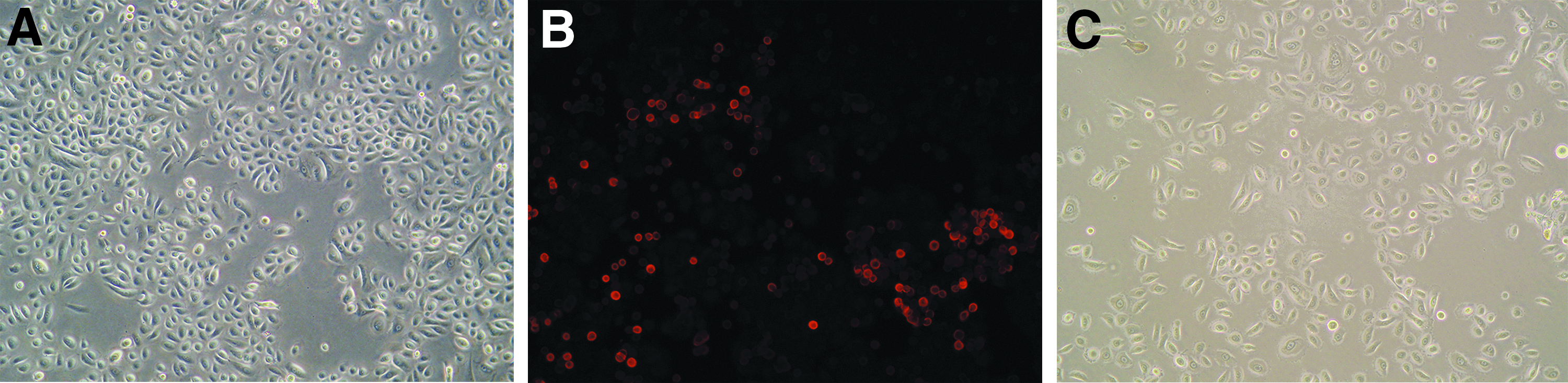
Immunohistochemical staining of those adherent cells with an antibody against K15 revealed a cytoplasmic staining pattern (Fig. 1B). Those cells displayed small and rounded shapes, with an even distribution on the bottom of the plate. They proliferated quickly and reached ∼40% confluence 4 days later, having shapes of round or small polygons or short shuttles (Fig. 1C), without contamination by fibroblasts.
Reprogramming of human epidermal keratinocytes
Keratinocytes at the second passage were transduced with a 1:1:1:1 mixture of retroviruses encoding four transcription factors: OCT4, SOX2, KLF4, and NANOG. The infection protocol consisted of two 45-minute infections with gentle shaking 24 hours apart. The infected cells were then seeded on a layer of irradiated MEFs (Fig. 2A), and within 4 days, several hundred tight cell colonies were observed (Fig. 2B). By day 10 post-infection, the typical morphology of iPSCs (tight colonies of cells with a large nuclear-to-cytoplasmic ratio) appeared (Fig. 2C), and by day 18 post-infection, some iPSC clones had grown large enough to be picked up to evaluate their characteristics (Fig. 2D).
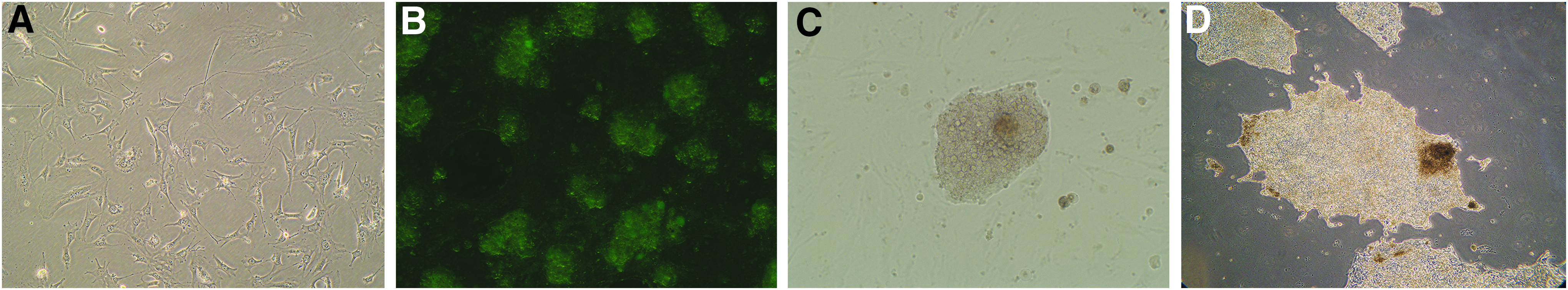
Characterization of irradiated MEFs and iPSCs.
Characteristics of acquired iPSCs
All established iPSC clones were positive for staining with antibodies against the stem cell markers SOX2 (Fig. 3A), OCT4 (Fig. 3B), NANOG (Fig. 3C), and SSEA3 (Fig. 3D) as well as staining with the CDy1 dye (Fig. 3E). All acquired iPSCs stained strongly positive for AP activity (Fig. 3F).
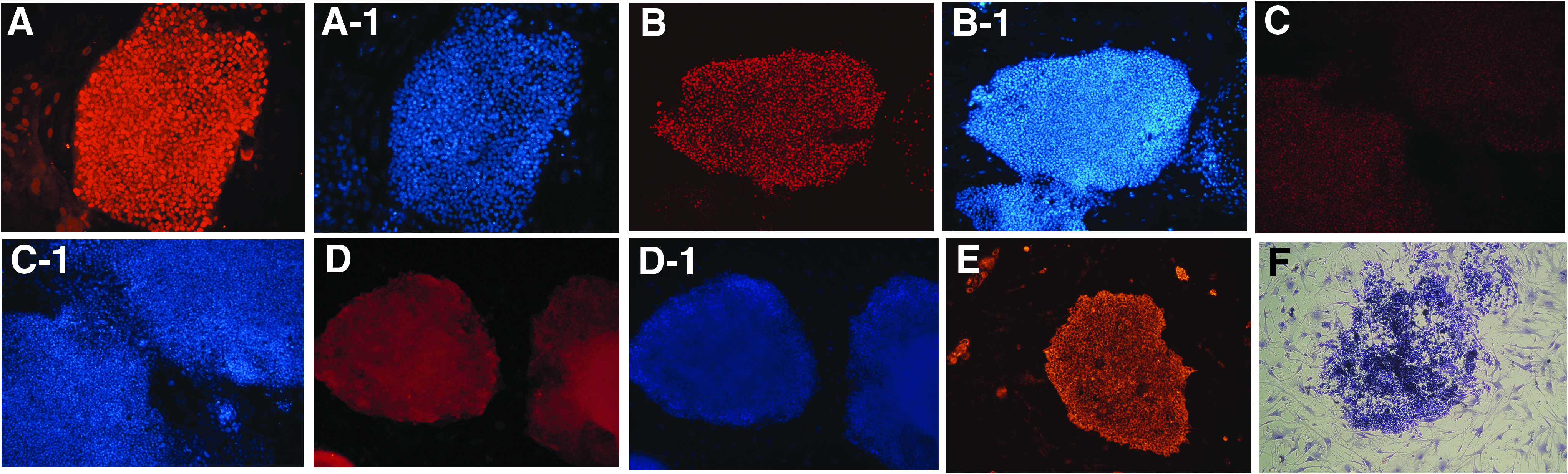
iPSCs were positive for staining with antibodies against the stem cell markers SOX2
Site-specific DNA methylation determination, namely Bisulfite sequencing, was used to measure the DNA methylation levels of the acquired iPSCs. The DNA methylation sites of acquired iPSCs and of primary epidermal keratinocytes were 23.8% (Fig. 4A) and 85% (Fig. 4B) in the OCT4 promoter region, and 42.5% (Fig. 4C) and 86.3% (Fig. 4D) in the NANOG promoter region, respectively. The difference in DNA methylation levels was obvious between the acquired iPSCs and the original primary keratinocytes. These results indicated that the iPSCs had lower levels of DNA methylation compared with their maternal cells.

Bisulfite sequencing of the OCT4
TEM analysis revealed that the iPSC cells had round nuclei with several nucleoli, scant cytoplasm (Fig. 5A), as well as poorly developed endoplasmic reticulum and Golgi complex (Fig. 5B), which is consistent with the general characteristics of stem cells.

TEM analysis showing iPSCs with round nuclei, each with several nucleoli, scant cytoplasm
Formation of EBs
EBs are three-dimensional aggregates formed in suspension by pluripotent stem cells, including ESCs and iPSCs. Plates coated with poly-HEMA and medium containing MC medium allowed the acquired iPSCs to grow in suspension. Two days after inoculation, many EBs began to form, and their volume gradually increased. On the sixth day of culture, most EBs had grown to a size that was appropriate for further adherent culture (Fig. 6A, B). The EBs were dispersed into individual cells to induce their differentiation into neuronal cells. In adherent cultures, the EBs quickly attached to the bottom of the gelatin-coated plates and grew well. After 1 week of culture, they stained strongly positive for OCT4 (Fig. 6C) and NANOG (Fig. 6D) and also stained with the CDy1 dye (Fig. 6E).

On day 6 of culture, most EBs had grown to a size that was appropriate for further adherent culture
In vitro differentiation
In addition to the multipotency of cell surface markers, one of the determining properties of iPSCs is to differentiate into three distinct germ lineages. To determine this characteristic, the first-generation clones were dispersed into individual iPSCs and were then treated with specific cytokines to induce adipocyte (mesodermal) and hepatocyte (endodermal) differentiation. The induction of iPSCs to adipocytes was confirmed by the production of oil droplets, positive for Oil Red O staining (Fig. 7A). The positive staining for AFP indicated the successful induction of hepatocytes (Fig. 7B). It usually took 3 weeks to produce adipocytes and hepatocytes from iPSCs. In contrast, inducing neurons took a longer time, ∼6 weeks. The successfully induced neuronal cells expressed Tubulin β-III and had a neuron-like morphology (Fig. 7C).

The induction of iPSCs to adipocytes was confirmed by the production of oil droplets that were positive for Oil Red O staining
Teratoma formation
The injection of acquired iPSCs gave rise to a 1–2 cm outgrowth on the mouse's hind leg at the site of injection within 4 weeks (Fig. 8A). The tumor gradually increased in size until week 8, when the mouse was sacrificed and the tumor with a diameter of 15 mm was harvested (Fig. 8B) for immunohistochemical analysis and cell culture. Histopathological examination of the tumor revealed the presence of all three germ layers (Fig. 8C). The tumor cells had large nuclei with obvious nucleoli and abundant cytoplasm. Immunohistochemistry showed positive staining for Vimentin (Fig. 9A) and CK(pan) (Fig. 9B) as brown-yellow particles in the cytoplasm, and for KI-67 (Fig. 9C) in the nucleus, but negative staining for GFAP (Fig. 9D) and AFP (Fig. 9E).
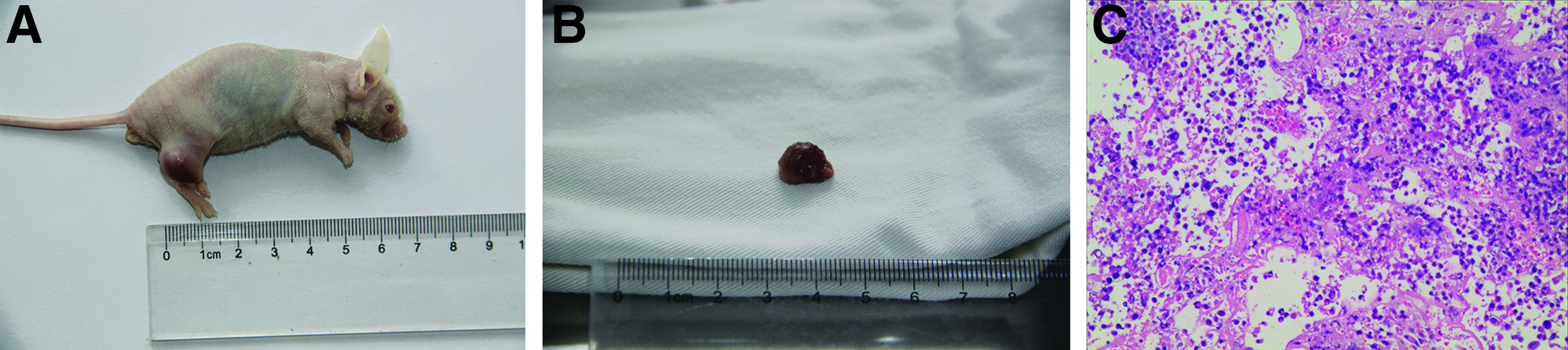
Within 4 weeks, the injection of iPSCs gave rise to a 1–2 cm outgrowth on the mouse's hind leg at the site of injection
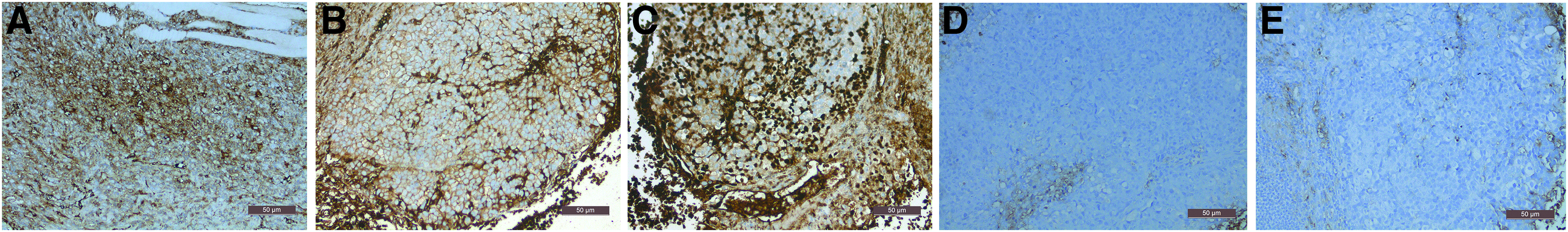
The tumor cells had large nuclei with obvious nucleoli and abundant cytoplasm. Immunohistochemistry indicated that Vimentin
One week after culture, cells from the teratoma tissue showed a spindle or oval shape when cultured in DMEM (Cat. No. SH30022.01B; Hyclone) (Fig. 10A, B), a closely arranged polygonal shape when cultured in KC medium (Fig. 10C, D), and a sparsely arranged long spindle shape when cultured in M254 medium (Cat. No. M-254-500; Life Technologies) (Fig. 10E, F). Interestingly, the tumor cells grew best in KC medium and maintained strong proliferation after 5 consecutive passages. Cells grown in KC medium were used for immunocytochemical analysis. Those results were consistent with the immunohistochemistry results of the tumor tissue, namely positive for Vimentin (Fig. 11A), CK(pan) (Fig. 11B), KI-67 (Fig. 11C), and OCT3/4 (Fig. 11D) but negative for GFAP (Fig. 11E) and AFP (Fig. 11F).

One week after culture, cells from the teratoma tissue showed a spindle or oval shape when cultured in DMEM
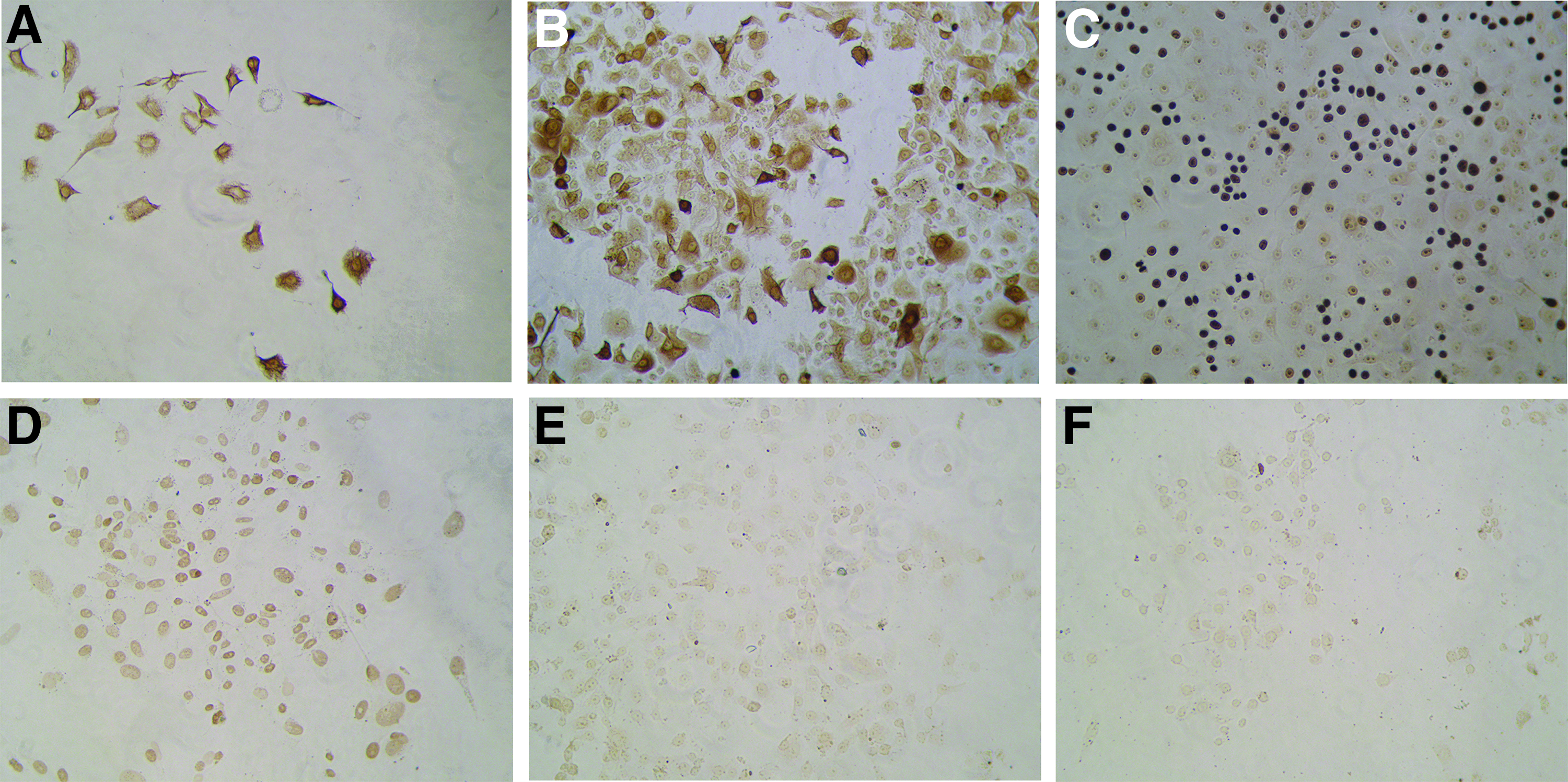
The teratoma cells grew optimally in KC medium and maintained strong proliferation after five consecutive passages. Cells grown in KC medium were used for immunocytochemistry. The results were consistent with the immunohistochemistry of the tumor tissue, namely positive for Vimentin
Discussion
The successful, rapid, and efficient generation of iPSCs could facilitate cell replacement therapy (Loring, 2018). However, the efficiency of existing reprogramming systems varies from 0.01% up to 1%, depending on the protocol used and the type of starting cell used. Transduction of somatic cells with four transcription factors (OCT4, SOX2, KLF4, and c-MYC) can generate fully reprogrammed iPSCs (Lorenzo et al., 2013). The type and property of the original cells used plays a pivotal role in the reprogramming process regarding the efficiency and timeframe. So, it is an important issue in iPSC generation to choose an appropriate source of cells to use. Skin cells are the most widely used types of cells in reprogramming systems because they are easily available, and fibroblasts are the most commonly used type of skin cell (Raab et al., 2014).
Recently, however, keratinocytes seem to be a more useful starting material for reprogramming since they can not only be obtained from routine outpatient minor surgeries but even be obtained by pulling hair follicles, processes that are minimally invasive or non-invasive. Many studies have shown that large numbers of keratinocytes can be obtained not only from routine outpatient minor surgeries but even by pulling hair follicles. The study of Aasen and Belmonte (2010) carefully described the isolation and culture of normal human epidermal keratinocytes from foreskin samples or punch biopsies and from plucked hairs.
Moreover, keratinocytes express higher basal levels of KLF4 and c-MYC, which may increase the efficiency of successful reprogramming, because the gene expression patterns of the donor cells play an important role in the reprogramming process. In 2008, Aasen et al. carried out the reprogramming of juvenile human primary keratinocytes into iPSCs with an approximately 100-fold greater efficiency and in half the time compared with the reprogramming of human fibroblasts.
In 2009, Hanna et al. reported that iPSCs preferentially arose from a particular epigenetic state in the donor cell population, such as progenitors or adult stem cells. In other words, progenitors or adult stem cells could be more easily reprogrammed to generate iPSCs, possibly due to the fewer stochastic steps required. Needless to say, if epidermal stem cells were reprogrammed, better results would be obtained. Recent studies indicate that epidermal stem cells exist in the basal layer of the epidermis and in the outer root sheath of hair follicles (Tao et al., 2018; Xue et al., 2018).
As we know, there are multipotent stem cells in the germinal layer near the basement membrane to ensure their constant proliferation and the integrity of human skin. In 2013, Yang et al. provided a simplified and effective method for obtaining epidermal stem cells, which adhered to type IV collagen within 15 minutes. This method separated the rapidly attaching stem cells from the slower adhering more differentiated keratinocytes. In our study, we obtained epidermal basal cells according to their method, and the population obtained was rich in stem cells. Immunostaining for K15 showed many strongly positive cells. To prevent those cells from losing their stemness that favored reprogramming, we used the second generation of keratinocytes (i.e., the first-selected stem cell-rich population) as the starting cells.
To prevent the keratinocytes from becoming senescent too early, we also paid great attention to detail during the process of culturing cells. First, we used special low-calcium medium formulations, which had another benefit in that after starting the reprogramming, medium with a normal calcium level is used whereby all uninfected keratinocytes reduce or stop proliferating, and only infected cells continue to proliferate, a prerequisite to reprogram them into stem cells.
Second, the cell passage was performed before the cells reached 80% confluence, because keratinocytes tend to differentiate into a senescent state when they become overconfluent. Third, we used accutase instead of trypsin to disperse the cells. Treatment with accutase, which contains proteolytic and collagenolytic enzymes, provides a gentler method of detachment. Over-digestion may cause damage to stem cells, including iPSCs.
Immunofluorescence staining, methylation assays (bisulfite genomic sequencing), EB formation, and teratoma formation assays are methods commonly used to prove the potential of the generated iPSCs to differentiate into all three germ layers and their pluripotency. It is worth mentioning that, in addition to using antibodies to stem cell-specific markers, we used the CDy1 dye for stem cell staining. The CDy1 dye can be used to stain and image ESCs, iPSCs, and various other types of stem cells. CDy1 is a fast and simple method to selectively stain live pluripotent stem cells for fluorescence microscopy and flow cytometry (Kang et al., 2011).
In the teratoma experiment, we mixed Matrigel with the cell suspension to increase the chance of tumor formation. Further, cells from the teratoma tissue were cultured in different culture media for 2 months, which showed their different phenotypic characteristics in those different media.
In fact, there have already been several studies that generated iPSCs from epidermal keratinocytes by using various techniques. Similar to those studies, we prepared iPSCs by transducing epidermal keratinocytes with four transcription factors (OCT4, SOX2, KLF4, and c-MYC).
However, our procedure still has some important differences and advances from them. The novelties of our study are as follows: (1) Since differentiated keratinocytes are resistant to reprogramming, it is critical to obtain as many keratinocytes as possible. We have provided a simplified and effective method for obtaining epidermal stem cells with the help of type IV collagen, because more undifferentiated keratinocytes will adhere to the type IV collagen within 15 minutes. (2) We used the CDy1 dye for stem cell staining, which can be used to stain and image ESCs, iPSCs, and various other types of stem cells. This makes our experimental results more convincing.
In conclusion, using epidermal basal cells (enriched in stem cells) as starting cells, the generation of iPSCs could be achieved with a high efficiency (∼0.1%) and a reduced timeframe (the first colony appeared on day 10 after transfection). This advance in the generation of iPSCs should prove important to the further development of cell replacement therapies.
Footnotes
Acknowledgments
The authors are very grateful to Professor V.J. Hearing for help with the English language editing. The work was supported by the National Natural Science Foundation of China (Grant No. 81673078) and the Science and Technology Foundation of Jiangsu (Grant No. BL2014036).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.