
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) remain a promising approach to target diseases with a loss of functional parenchyma. This technology comes with a number of concerns for clinical applications, including teratogenic potential and genomic instability. Here we focused on evaluating the safety of cross-species Sendai viral reprogramming, as well as investigating the transcriptional dynamics during reprogramming and differentiation. We established that Sendai viral vectors carrying human Oct4, Sox2, Klf4, and c-Myc (OSKM) could produce mouse iPSCs free of transduced viral materials. Gene expression analysis revealed an efficient silencing of the virally-introduced human pluripotency factors and upregulation of the endogenous pluripotency network over time. In addition, single cell gene expression analysis of proof-of-principle–derived cardiomyocytes revealed distinct expression patterns indicative of subspecialized cardiac cell lineages. Moreover, our results demonstrate the importance of monitoring genomic aberrations before any clinical or preclinical applications, as we detected a high prevalence of chromosomal instability. Taken together, we demonstrated the successful use of a clinically germane method to reprogram terminally differentiated mouse cells and their potential to generate specialized cardiac cell types. Additionally, our results suggest a plasticity of OSKM to reprogram more divergent species and provide a new application of an established reprogramming approach.
Introduction
The advent of induced pluripotent stem cell (iPSC) technology has unlocked a powerful tool for the generation of large-scale disease models, drug screening libraries, and cellular transplantation therapies (Robinton and Daley, 2012). Since the seminal publication describing the generation of iPSCs from terminally differentiated cells (Takahashi and Yamanaka, 2006), scientists have sought to create safe approaches that would be suitable for clinical applications. To translate findings from animal models to the bedside, it is of utmost importance to approximate clinical approaches as closely as possible. The use of human-derived transplant material in animal models may obscure physiologic responses due to cross-species rejection and the requirement of continuous immunosuppression.
Cross-species physiological differences, such as heart beat frequency, may also create additional barriers for xenograft models. For these reasons, species-specific models may be advantageous over human xenograft systems. To this end, we established murine iPSCs using human factors and nonintegrating viral vectors. We provide a proof of concept in utilizing the mouse model as a test bench for clinical applications of iPSC technology by using the explicit reprogramming approach in human cells and downstream clinical treatments.
Multiple viral and molecular vectors (González et al., 2011; Schlaeger et al., 2015) are available for cell reprogramming; however, most have serious drawbacks, including host genome integration, selective infection of dividing cells, or a reprogramming efficiency too low to be of any clinical significance (Rao and Malik, 2012). The use of Sendai viral vectors to deliver OCT4, SOX2, KLF4, and c-MYC (OSKM) transgenes is perceived as a promising and efficient means to safely and reliably generate iPSCs for research and clinical applications (Ban et al., 2011).
Sendai viruses are nonintegrating, single-stranded RNA viruses that can infect cells independently of cell cycle (Fusaki et al., 2009; Li et al., 2000). Nonreplicative versions of this virus have been generated for OSKM delivery, and this anti-sense RNA virus remains cytoplasmic throughout viral infection, curbing the potential for integration of delivered transgenes (Fujie et al., 2014).
Sendai viral vectors carrying human OSKM have been previously used to generate iPSCs from human and chimp cells (Fujie et al., 2014; MacArthur et al., 2012). In this study, we explore the use of Sendai viral vectors with human OSKM to reprogram primary mouse cells. We demonstrated that Sendai-delivered human OSKM successfully generated pluripotent mouse iPSCs. Moreover, the use of human OSKM to reprogram mouse cells enabled the examination of pluripotency gene expression dynamics during reprograming. We observed an induction of endogenous mouse pluripotency genes, while the levels of Sendai viral genome and exogenous gene expression decreased over time.
Furthermore, single-cell analysis revealed that the generated iPSCs had the capacity to differentiate into various cardiomyocyte lineages, addressing the challenge of generating subspecialized cell types for downstream clinical applications. Our results provide novel insight for the capacity of OSKM in interspecies reprogramming. Additionally our results highlight the importance of monitoring for genomic aberrations as, while we successfully could generate pluripotent and normal mouse iPSCs, a high degree of genomic instability was detected. Ultimately, however, we propose that Sendai-delivered human OSKM, a clinically-relevant reprogramming approach, can create safe, transgene-free, and species-specific iPSCs for utilization in disease models or transplantation studies.
Materials and Methods
Animals and ethics statement
Hearts from adult wild-type C57/BL6JBomBmsd (Charles River) and αMHC-mCherry mice (a gift from Professor Bernd Fleischmann) were used to generate primary cardiac explants (Hesse et al., 2012). All animals were housed and treated in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animals with prior approval from the Institutional Animal Care and Use Committee. In facilities running 12/12 light/dark cycles at temperatures of 22°C–23°C, all mice had uninterrupted availability of both food and water. Animals were euthanized with carbon dioxide and immediate cervical dislocation.
Fibroblast isolation
Fibroblasts from adult cardiac explants were obtained as previously described (Qian et al., 2013). Briefly, hearts were isolated, washed with sterile phosphate-buffered saline (PBS), and minced in explant media [Iscove's modified Dulbecco's medium (IMDM), 20% fetal bovine serum (FBS), 1% penicillin-streptomycin]. Explant pieces were resuspended in explant media and placed in 0.1% gelatin-coated dishes. Tissue was allowed to settle for 2 hours at 37°C in 5% CO2, upon which additional media was added. The explants were not disturbed for an initial 3-day settling period, after which the media was changed as needed. Before expansion, cells were filtered through a 100 μm mesh.
Reprogramming and cell maintenance
Within the first three passages from isolation, fibroblasts in basic fibroblast media (Dulbecco's modified Eagle's medium [DMEM] + GlutaMAX, 10% embryonic stem cell (ESC)-qualified FBS, 0.1 mM nonessential amino acids, 55 μM β-mercaptoethanol) at 80%–90% confluence were transduced with CytoTune 2.0 Sendai Virus (ThermoFisher) at a multiplicity of infection (MOI) of 5 for each viral vector (KOS, Klf4, and c-Myc). Fibroblast media was changed the day after transduction and every other subsequent day. Seven days after transduction, cells were transferred to mouse embryonic fibroblast feeders that were previously inactivated with mitomyocin C treatment.
One day after transfer, cells were cultured in mouse embryonic stem cell media [Knock-out DMEM, 15% Knock-out Serum Replacement, 0.1 mM nonessential amino acids, 1% GlutaMAX, 55 μM β-mercaptoethanol, and 10 ng/mL mouse leukemia inhibitory factor (mLIF)], and the media was changed every following day with fresh mouse embryonic stem cell media. Colonies were observed and picked 11–13 days after initial transduction. Clones were maintained on feeders in mouse embryonic stem cell media and passaged at varying densities within 24–48 hours of each other using Accutase. All clonal lines were frozen between passage 6 and 8 and thawed for subsequent passaging. The clonal line that was passaged beyond passage 8 went through an additional freeze-thaw cycle. Freeze-thaw cycles were not considered passaging of cell lines.
RNA isolation, complementary DNA production, and preamplification
For quantitative real-time polymerase chain reaction (qPCR) analysis of reprogrammed bulk populations, the FastLane Cell cDNA Kit (Qiagen) was used for RNA isolation and complementary DNA (cDNA) production. Approximately 40,000 cells, or one confluent 24-well plate, were processed according to the manufacturer's protocol. Briefly, cells were lysed in 200 μL lysis buffer and the cell lysate was frozen at −80°C overnight for increased lysis before cDNA production. For cDNA production, 4 μL cell lysate was mixed with 2 μL gDNA wipe-out buffer and 8 μL water. This was incubated for 5 minutes at 42°C, placed on ice, and mixed with 1 μL Quantiscript reverse transcriptase, 1 μL RT Primer Mix, and 4 μL Quantiscript RT Buffer, followed by incubation for 30 minutes at 42°C and 3 minutes at 95°C.
The preamplification of bulk populations was performed according to the Gene Expression PreAmp protocol provided by Fluidigm and using the Fluidigm PreAmp Master Mix. Pooled 0.2 × assay mix was prepared in advance by diluting 1 μL of each 20 × TaqMan Gene Expression Assay (Applied Biosystems) to a total volume of 100 μL Tris-EDTA (TE)-buffer. The preamplification reaction of each sample was prepared with 1 μL PreAmp Master Mix, 1.25 μL Pooled TaqMan assay mix (0.2 × ), 1.5 μL water, and 1.25 μL cDNA.
Samples were preamplified with the following thermocycle protocol: 50°C for 900 seconds and 95°C for 120 seconds, followed by 95°C for 15 seconds and 60°C for 240 seconds for 14 cycles. Bulk population samples were diluted 4 × with TE-buffer before storage at −20°C, and preamplified cDNA was utilized at a dilution of 1:10 when analyzed on the BioMark.
For isolated single-cell qPCR gene expression assays, single cells were sorted into 96-well plates prepared in advance with a cell lysis and preamplification mixture as previously described (Tarnawski et al., 2015). Thermocycling protocol for preamplification was as described for bulk populations with the exception of 20 total cycles used for single cells. Preamplified cDNA was diluted 5 × with TE-buffer and analyzed on the BioMark. The cDNA production and preamplification for both single cells and bulk populations were performed using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad).
BioMark Real-Time PCR Fluidigm™
All qPCRs were performed using the high-throughput BioMark Real-Time PCR System (Fluidigm, South San Francisco, CA) as previously described (Tarnawski et al., 2015). Nontemplate controls as well as a sample without reverse transcription were included with the purpose of detecting possible background amplification. Positive controls from mouse and human iPSCs, as well as BioBank generic pooled cDNA (Primer Design, United Kingdom), were included for verification of the assays. The specific hydrolysis probes used for cDNA amplification and qPCR experiments can be found in Supplementary Table S1.
Assays for cytogenetic stability and pluripotency
Cells free of detectable levels of Sendai virus were used for fluorescence in situ hybridization (FISH), karyotypic, and teratoma analyses. All samples used for these analyses underwent a single round of feeder depletion in culture before downstream testing. Feeder depletion involved a single round of passaging onto gelatin-coated dishes while in the presence of adaptation media (IMDM, 20% knockout serum replacement, 1 × GlutaMax, 150 μM monothioglycerol, and 10 ng/mL mLIF).
FISH analysis and karyotyping
FISH was performed as previously described (VanBrocklin et al., 2009). Probes used for hybridization were created with bacterial artificial chromosome (BAC) clones RP23–151l21 (Chr 8A1.1) and RP23–314K19 (Chr 11B2) (BACPAC Resource Center). Purified BAC clones were labeled by nick translation with Green-dUTP and Orange-dUTP (Abbott Molecular Incorporated). For FISH analysis, FISHView v7.2 software (Applied Spectral Imaging [ASI]) was used and 200 cells were scored for each sample.
Spectral karyotyping (SKY) was also performed as previously described (VanBrocklin et al., 2009). In brief, denatured metaphase slides were incubated with denatured SkyPaint probe (ASI). Slides were then covered with glass coverslips and incubated overnight in a 37°C humidified chamber. Washes for pretreatment and posthybridization were completed following the standard supplied protocol (ASI) with slight modifications. An Olympus BX43 microscope was utilized to view slides, and image acquisition was performed at either 600 × or 1000 × magnification. Images were taken using a COOL-1300 SpectraCube camera outfitted with a SKY optical filter (ASI). At least 20 metaphases were analyzed for each sample using the HiSKY v7.2 software (ASI).
For reverse 4′,6-diamidino-2-phenylindole (DAPI) karyotyping, metaphase slides were incubated at 55°C overnight, followed with the application of DAPI. FISHView analysis software (ASI) was used to visualize inverted DAPI images. At least 20 metaphases were analyzed for each sample.
Teratoma assay
Athymic nude mice were subcutaneously injected with 106 iPSCs resuspended in 100 μL DMEM and 10% FBS. As a positive control for tumor formation, 106 HT1080 human fibrosarcoma cells were injected in parallel. Tumors were monitored and harvested at or before the maximum endpoint volume of 1500 mm3. Isolated tumors were fixed in 4% paraformaldehyde (PFA) at 4°C and paraffin-embedded after equilibration to 70% ethanol. Sections were stained with hematoxylin and eosin, and tissues were identified by a trained pathologist.
In vitro differentiation
To generate embryoid bodies (EBs) for immunofluorescence, iPSCs were transferred to low-attachment culture dishes in leukemia inhibitory factor (LIF)-free mouse embryonic stem cell media (Knock-out DMEM, 15% Knock-out Serum Replacement, 0.1 mM nonessential amino acids, 1% GlutaMAX, 55 μM β-mercaptoethanol) and cultured for 3 days with slow rocking. Aggregated cells were plated onto 0.1% gelatin-coated dishes and incubated for an additional 3 days. Media change occurred each day after plating onto gelatin-coated dishes.
For cardiac-specific differentiation, a previously described protocol was followed with slight adaptations (Kattman et al., 2011). In brief, miPSCs were maintained on feeders in mouse embryonic stem cell media. Before differentiation, feeders were depleted with one passage on 0.1% gelatin-coated dishes in adaptation media. Confluent plates were resuspended using Accutase, and cells were cultured at 1.5 × 105 cells/mL in serum-free differentiation media (75% IMDM, 25% Ham's F12 media, 0.5 × N2, 0.5 × B27 [without retinoic acid], 0.05% bovine serum albumin, 2 mM glutamine, 0.5 mM ascorbic acid, 100 U/mL penicillin streptomycin, and 4.5 × 10−4 M monothioglycerol) for 48 hours with continuous and gentle rocking.
Aggregated cells were then dissociated with Accutase and re-aggregated in serum-free differentiation media with the addition of activin A (8 ng/mL), BMP4 (0.5 ng/mL), and VEGF (5 ng/mL). Cells were re-aggregated in 10 cm low-attachment culture dishes with gentle rocking for ∼30 hours. Cells were then incubated with Accutase and replated on gelatin in modified cardiac condition media (StemPro-34, 2 mM glutamine, 1 mM ascorbic acid, 5 mM human VEGF [&D Systems], 150 ng/mL human DKK1 [R&D Systems], 10 ng/mL human bFGF [Life Technologies], and 12.5 ng/mL human FGF10 [R&D Systems]). Media was changed as needed, and cells were harvested after a total of 9 days of differentiation.
Fluorescence-activated cell sorting (FACS)
Differentiated iPSCs were dissociated using AccuMAX and resuspended in basic maintenance media (DMEM, 15% ESC-qualified FBS, and 100 U/mL penicillin-streptomycin) before filtration through a 70 μm mesh. Cells were stained with DAPI (2 μg/mL) and were sorted on a BD Biosciences Influx using a 100 μm nozzle. Single and unstained controls were used to set gates.
Immunofluorescence
Cells and EBs were fixed at 4°C with 4% paraformaldehyde, permeabilized in PBS/0.1% Triton X-100/0.1% Tween20, blocked with 10% goat or donkey serum (Sigma-Aldrich), and stained with primary antibodies: goat anti-Oct4 (Abcam), rabbit anti-Nanog (Abcam), mouse anti-SSEA1 (Abcam), rabbit anti-Brachyury T (Abcam), rabbit anti-Foxa2 (Abcam), and rabbit anti-Sox1 (Cell Signaling Technology). Primary antibodies were visualized with secondary antibodies conjugated to Cy3/Cy5 (Jackson ImmunoResearch) or Alexa Fluor 555/647 (Invitrogen). DAPI (Invitrogen) was used to visualize nuclei. Imaging was performed using Leica DM500 B (Switzerland).
Statistical analysis
Gene expression data from the reprogramming time course study were generated on the Fluidigm BioMark HD platform. Results were exported in the standard output format for subsequent analysis. Serial dilutions were run for each primer set, and primer efficiencies were estimated using techniques previously described (Pfaffl, 2001). Estimated efficiencies were explored graphically; poorly performing primers with median efficiencies not falling within a range of 100% ± 20% were excluded from further analysis. Gapdh and Sdha were selected as reference genes following the approach described in Vandesompele et al. (2002) for the bulk analysis in Figure 1.

Gene expression dynamics and viral kinetics throughout reprogramming of primary mouse cells with human OSKM delivered by Sendai viral vectors.
Efficiency-corrected relative expression ratios for all time points were computed relative to samples taken 24 hours after transduction with Sendai virus. Mean and standard deviation values for all replicates at each time point are plotted in Figure 1. All analyses were performed using the R programming language (v3.2.3) (RDC Team, 2015). Sendai viral expression was normalized to Gapdh expression, and the qPCR data were analyzed using the GenEx software (Multid). For Figure 1C, a two-sided t-test was used and a p value <0.05 was considered statistically significant.
Results
Reprogramming using human OKSM factors upregulated the endogenous mouse pluripotency network
Cardiac tissue explants from adult mice (n = 3) were expanded and transduced with recombinant Sendai viral vectors encoding human KLF4, OCT4, SOX2, and c-MYC (OKSM) (Fig. 1A). To study the gene expression dynamics during Sendai viral reprograming of mouse cells using human factors, we preformed species-specific qPCR analysis of cells and iPSC clones derived from these primary mouse cardiac tissue explants. The expression of core endogenous pluripotency network genes, specifically Sox2, Oct4, and Nanog, prominently increased between days 7 and 11 post-transduction and plateaued within the first passage of heterogeneous populations (Fig. 1B). By passage 6, a 53-, 29-, and 36-fold increase was detected in murine Oct4, Nanog, and Sox2 expression, respectively.
Notably, a twofold decrease in endogenous c-Myc was observed by day 7 and remained at this level of expression throughout the analysis (Fig. 1B). In contrast, Klf4, which is thought to repress somatic genes early during reprogramming and induce the expression of pluripotency genes in the later hierarchical phase (Polo et al., 2012), demonstrated a twofold increase in expression that plateaued by passage 4. In parallel, levels of exogenous human KLF4 and Sendai viral genomes decreased ∼900-fold and 35-fold, respectively, by passage 4 (Fig. 1B). Human SOX2 and OCT4 were not detected in transduced mouse cells. However, the expression of these genes was observed in control human iPSCs (Fig. 1C), confirming the validity of the assay.
During early reprograming, the analysis of fibroblast-associated genes identified a significant decrease in expression of β-actin, fibronectin 1, and vimentin, consistent with a loss of fibroblast identity (Fig. 1D) (Mikkelsen et al., 2008). In addition, a significant increase in expression of stem cell antigen 1 (Sca1), a progenitor cell marker (Holmes and Stanford, 2007), was observed within 11 days of transduction (Fig. 1D).
Generated iPSCs are pluripotent
Pluripotency-associated gene expression was further characterized at the protein level using immunofluorescent staining. Heterogeneous and isolated clonal populations appeared to have tight colonies and distinct borders typical of iPSC induction (Fig. 2A). Protein expression of SSEA1, an early maker of iPSCs, was confirmed (Hansson et al., 2012; Polo et al., 2012). Furthermore, nuclear transcription factors of established pluripotency, including NANOG and OCT4 (Fig. 2B, C), were also expressed in reprogrammed clones (Mikkelsen et al., 2008).
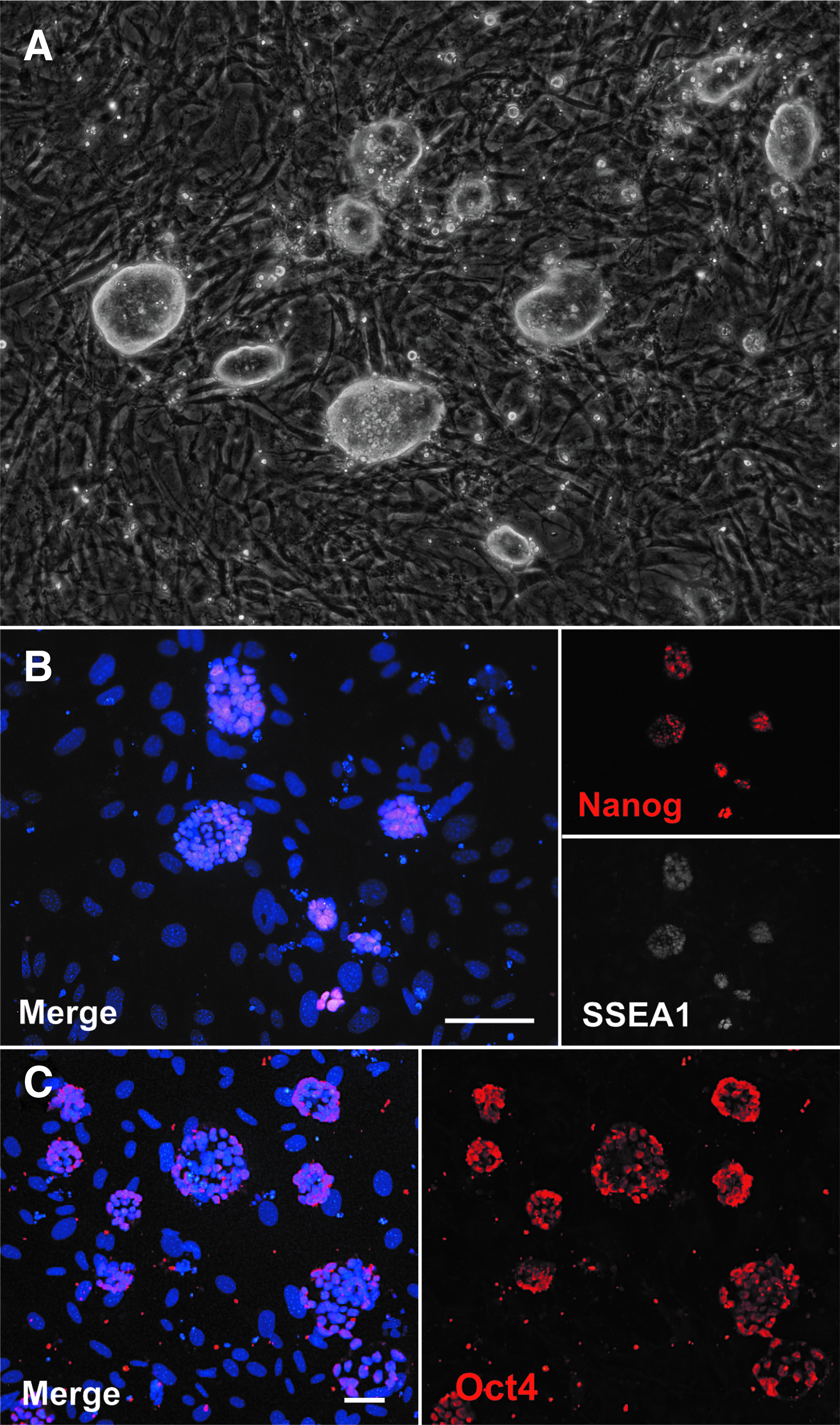
Immunofluorescence of generated mouse iPSC clones reveals the expression of pluripotency genes.
To further characterize generated clones and verify pluripotent potential, clonal iPSC lines were differentiated in vitro using a modified mass culture method (Takahashi and Yamanaka, 2006). Generated EBs were positive for markers of all three embryonic germ layers such as SOX1 (ectoderm), FOXA2 (endoderm), BRACHYURY T (mesoderm), and GATA4 (meso-endoderm) (Fig. 3A–D). Furthermore, iPSCs were injected into nude mice for the assessment of teratoma formation. Two of three investigated lines produced tissues originating from the three developmental germ layers, including ectoderm (squamous epithelium, immature neuro-ectoderm, and hair follicles), endoderm (glandular epithelium), and mesoderm (adipose and muscle tissue) (Fig. 3E–J).
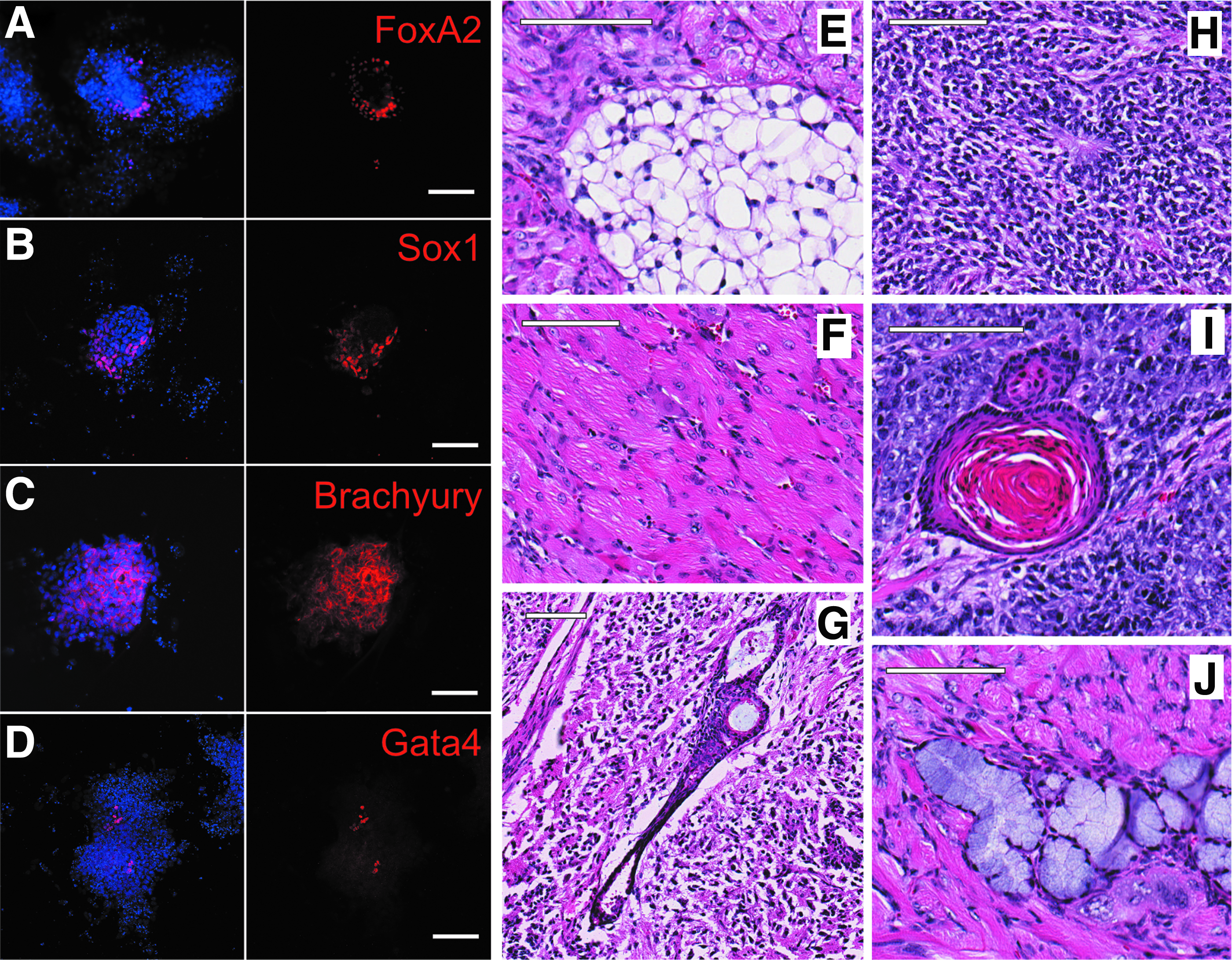
Differentiation potential of generated mouse iPSCs. Immunofluorescence of in vitro differentiated mouse iPSCs confirms the expression of
Generated iPSCs are free from viral transcripts but cytogenetically unstable
To evaluate the safety of the reprogrammed material, we performed a more detailed examination of Sendai viral genomes in passaged clones using qPCR. Eight out of nine clones lost detectable levels of the virus by passage 8 (Fig. 4A). Additionally, expression of human KLF4 was lost at the time of, or before, the loss of Sendai viral genomes (Fig. 4B). Taking advantage of the temperature-sensitive nature of the viral genome, the single clonal line with detectable levels of the virus at passage 8 was incubated at 39°C at the start of passage 10. By the end of passage 10, this clonal line lost detectable levels of the virus through standard qPCR and could no longer be detected with additional preamplification by passage 12 (data not shown).

Presence of Sendai virus and expression of human KLF4 during reprogramming. Presence of
As the reprogramming process involves an increase in replication stress that is thought to give rise to genomic instability (Ruiz et al., 2015), we sought to investigate the genomic integrity of Sendai virus-generated iPSCs using FISH, reverse DAPI karyotyping, and/or SKY (Table 1 and Supplementary Table S2). Detailed analysis revealed significant abnormalities, including translocations, deletions, and aneuploidy, in five of the six clones. One clone (A4) appeared cytogenetically normal when analyzed with reverse DAPI and SKY (Fig. 5A, B). This clone also showed pointedly lower levels of individual cells exhibiting polyploidy in culture when analyzed with FISH (Fig. 5C and Supplementary Table S2).

Cytogenetic analysis of pluripotent cell lines (n = 6). Representative reverse DAPI
Composite summary of karyotype data from generated iPSCs
Ultimately, these results suggest that reprogramming of primary mouse cells with human OSKM and Sendai viral delivery can produce cytogenetically normal and pluripotent mouse iPSCs.
Generated iPSCs have cardiac differentiation potential and can generate chamber-specific and pacemaker-like cardiomyocytes
To look more deeply into mesodermal differentiation potential and to establish whether generated iPSC lines could differentiate into multiple lineages of the heart, we differentiated iPSCs derived from the αMHC-mCherry transgenic mouse line (cytogenetically normal clonal line A4). In these mice, cells that have committed to a working cardiomyocyte fate will express mCherry from an alpha-myosin heavy chain promoter and be detectable by immunofluorescence (Hesse et al., 2012). This generated iPSC line was differentiated using a modified, previously published method (Kattman et al., 2011) (Fig. 6A). αMHC-mCherry-positive cardiomyocytes were observed during differentiation (Fig. 6B) and corresponded to beating areas within the dish.
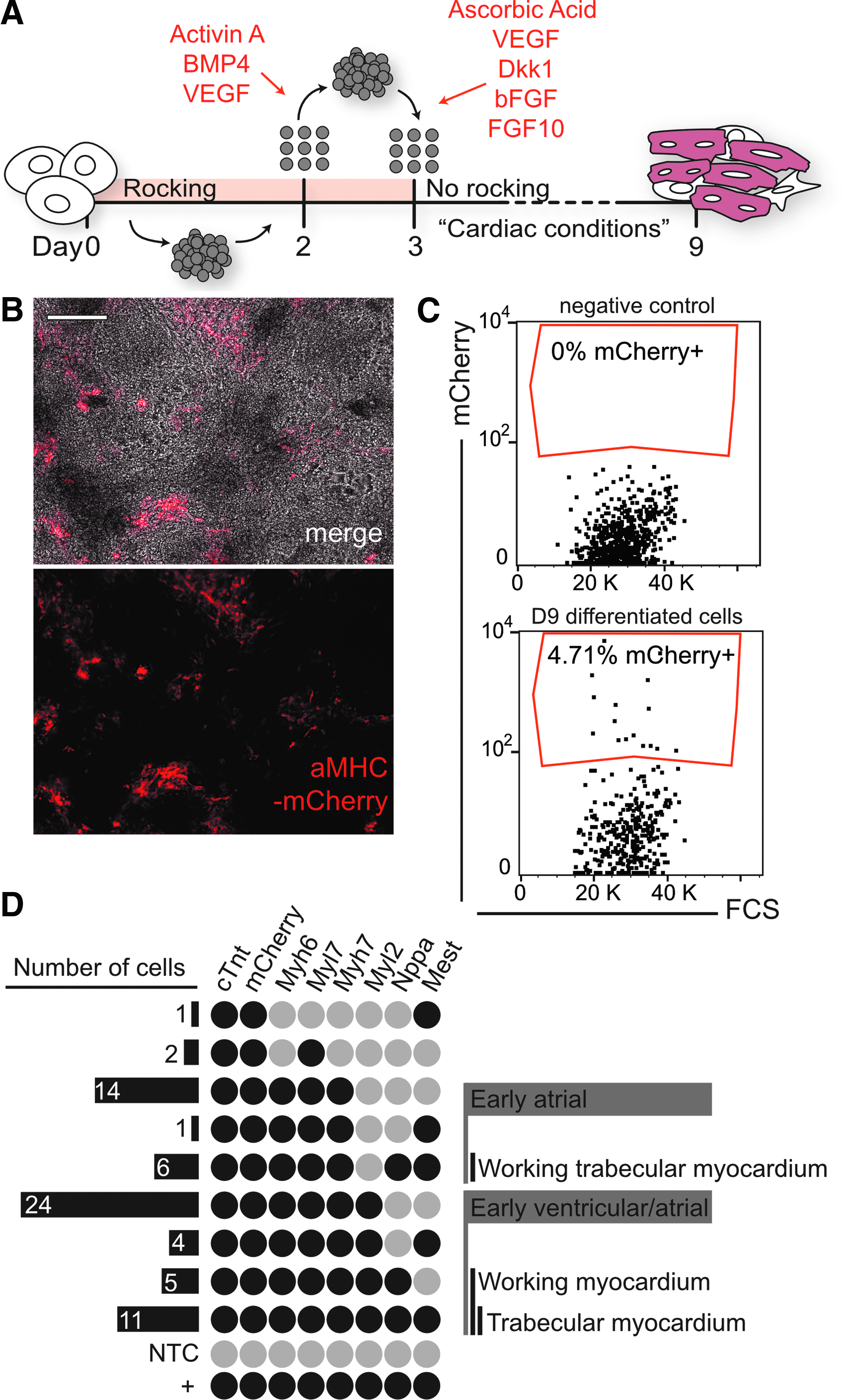
Cardiac differentiation potential of generated mouse iPSCs.
After 9 days of differentiation, 4.71% were positive for mCherry, as analyzed with flow cytometry (Fig. 6C). αMHC-mCherry-positive cardiomyocytes were isolated for single-cell gene expression analysis of cardiac-associated genes (Fig. 6D).
During mouse development, subpopulations of cardiomyocytes express varying patterns of heavy and light chain myosins, providing gene expression profiles that can differentiate between cardiomyocytes of various chambers. At late stages of mouse development, myosin light chain 2v (Myl2) expression is limited to the ventricles and the outflow tract (Moorman and Christoffels, 2003), while atrial cardiomyocytes express high levels of myosin light chain 2A (Myl7) (Small and Krieg, 2004; Zammit et al., 2000). In addition, myosin heavy chain 6 (Myh6) is expressed at higher levels in the atria during murine development, while myosin heavy chain 7 (Myh7) is found restricted to ventricular cardiomyocytes beyond day 9 of mouse embryogenesis (Lyons et al., 1990).
Therefore, cardiomyocytes expressing high levels of Myl2 and Myh7 would most likely represent ventricular cardiomyocytes, while cells expressing high levels of Myl7 and Myh6 may delineate cardiomyocytes of the atria. Early cardiomyocytes can also be more acutely classified as constituents of the compact or trabecular myocardium, the latter expressing significantly higher levels of mesoderm-specific transcript (Mest) (King et al., 2002). In addition, atrial natriuretic factor (Nppa) expression demarcates the atrial and trabecular myocardium during rodent development (Christoffels et al., 2000).
These markers for various cardiomyocyte populations were examined within our isolated mCherry-positive single cells. From 81 sorted single cells expressing Gapdh, single-cell qPCR analysis revealed that 72 were positive for mCherry and 77 expressed cardiac troponin T (cTropT), a general marker for cardiomyocytes. An early atrial population (Myl2−) was observed, as well as an early atrial population exhibiting markers for working trabecular myocardium (Mest+ and Nppa+) (Fig. 6D). A potential group of immature cardiomyocytes was observed (Myl2+, Myh7+, Myl7+, Myh6+).
The expression prolife of these cells may indicate an early ventricular but also early atrial origin as they have not lost the expression of Myl2 or Myh7. Within this developing population, working trabecular-like (Mest+ and Nppa+) and possible atrial cardiomyocytes (Nppa+) were observed. When examining a clustered heat map of gene expression data, frequent co-expression of cardiac Myl2, Myl7, cTropT, Myh6, Myh7, and mCherry was observed. A similar clustering of trabecular working myocardial Nppa and Mest expression was also identified, as calculated by Ward's algorithm with an Euclidean distance (Supplementary Fig. S1). In addition to the working myocardium of both chambers, subpopulations of potential pacemaker-like cells were also observed within this analysis.
Five single cells expressed Hcn4, Tbx18, and Tbx3 (Supplementary Fig. S1), a combination of transcription-associated factors that are characteristic of sinoatrial cells, while 18 cells lacking the expression of Tbx18 expressed Hcn4 and Tbx3 (Supplementary Fig. S1), an expression pattern delineating pacemaker cells of the atrioventricular node (Christoffels and Moorman, 2009). Furthermore, these pacemaker cells (Tbx18, Tbx3, and Hcn4) showed a high degree of similarity in the hierarchical clustering analysis (Supplementary Fig. S1). These results not only highlight the potential of the derived iPSCs to generate many different cell types during differentiation, but also demonstrate the capacity of these iPSCs to generate distinct subtypes of cardiomyocytes that could be used for targeted and specific therapeutic approaches.
Discussion
Our report is the first to successfully demonstrate the use of viral preparations intended for human cells in the reprogramming of primary mouse cells. What is more, we provide important insight into transduced OSKM dynamics by utilizing the inherent divergence of human and mouse OSKM to track ectopic and endogenous gene expression throughout the reprogramming process.
Upon the introduction of OSKM-encoding Sendai virus, we observed an immediate decrease in the expression of terminally-differentiated fibroblast markers, including vimentin and fibronectin 1, as well as the cytoskeletal gene beta actin. Together with a rapid increase in the expression of the progenitor marker Sca1 (Holmes and Stanford, 2007), these results reflect the early stages of de-differentiation previously reported during reprogramming in mouse (Mikkelsen et al., 2008). With delayed onset, which is temporally consistent with a late, hierarchical phase of reprogramming (Buganim et al., 2012), there is a robust increase in the expression of endogenous Oct4, Sox2, and Nanog (Fig. 1B).
Taken together, these results recapitulate the temporal dynamics seen in retroviral reprogramming of mouse cells using murine OSKM (Mikkelsen et al., 2008; Polo et al., 2012). Moreover, this would indicate that the biphasic model previously described (Mikkelsen et al., 2008; Polo et al., 2012) can be attributed to the reprogramming process in general and not to the method of factor delivery. When endogenous Oct4 and Sox2 reached a plateau of expression within the first two passages, a concurrent decrease of transduced human KLF4 was observed (Fig. 1B). Interestingly, an equally robust change in the endogenous levels of Klf4 or c-Myc could not be detected. Klf4 and c-Myc are thought to drive the first wave of changes during reprogramming characterized by an increased expression of proliferation-associated genes (Polo et al., 2012).
This could indicate that the human-derived genes are enough to spark de-differentiation, reducing the need for endogenous activation. This mechanism could be beneficial, as minimal to no induction of endogenous Klf4 and c-Myc expression may promote cell populations that would undergo less replicative stress and result in lower levels of genomic instability in iPSCs (Ruiz et al., 2015).
In our hands, five out of six clonal iPSC lines analyzed exhibited karyotypic abnormalities, including major deletions and translocations (Table 1). This rate of genomic instability is higher than previously reported, as 38% and 22.8% of mouse ESC and iPSC lines, respectively, were reported to have abnormal karyotypes, and <13% of human ESC or iPSC lines had detectable cytogenetic abnormalities (Ben-David and Benvenisty, 2012; Taapken et al., 2011). More investigation is required to determine if this apparent increase in cytogenetic instability is due to Sendai viral delivery of OSKM, or if it may be due to the use of human OSKM to reprogram primary mouse cells. Furthermore, it would be valuable to determine if nucleoside supplementation would increase genomic stability in this context as previously described (Ruiz et al., 2015).
Our results do emphasize, even with any additional methods aimed to reduce the frequency of karyotype aberrations, the need for high-throughput monitoring of genomic aberrations or more stringent whole-genome sequencing before downstream clinical applications.
In addition to investigating ectopic and endogenous gene expression, levels of Sendai virus were observed throughout reprogramming. Sendai virus levels were stable at the early stages of reprogramming and dropped precipitously upon clonal passaging (Figs. 1B and 4A). Mirroring previous accounts of Sendai viral genome loss during reprogramming (Fujie et al., 2014; MacArthur et al., 2012), all but one clonal line analyzed lost detectable levels of the virus by passage 8, and the remaining clonal line quickly lost the viral genome upon passaging at nonpermissive temperatures (data not shown).
As wild-type Sendai virus is found endemically in rodents (Parker et al., 1978) and co-infection with recombinant virus could result in the rescue of replication deficiency found in the recombinant version, it is of utmost importance to screen potential iPSC lines for the presence of viral genomes before their use in animal models.
Our results indicate that, while the majority of generated iPSC lines lose the virus and exogenously introduced human gene expression by passage 8, loss of the virus in culture is variable, and all material that is planned for further use in downstream applications should always be tested for the presence of Sendai viral genomes. Importantly, heating the cells to 39°C guaranteed the loss of viral vectors and OSKM factor genes.
Although previous reports indicate that human OSKM can revert terminally differentiated human, chimp, and porcine cells to a pluripotent state, it was unclear if cells from murine origin would be too divergent to respond in a similar fashion (Ezashi et al., 2009; Fujie et al., 2014; Schlaeger et al., 2015). In vitro differentiation of potential mouse iPSCs resulted in cells positive for markers of all three embryonic germ layers, and the injection of three clonal iPSC lines resulted in sizable teratomas from two clonal lines (lines 2.1 and A4). Interestingly, the clonal line that did not produce sizable teratomas (line 2.5) had the most abnormal karyotype. This may have prevented this line from differentiating properly into tissues of the three developmental germ layers, indicating that karyotype changes may counteract pluripotency rather than promote it.
The practical application of mouse iPSCs generated through human factors was more thoroughly investigated by mesodermal differentiation of clonal iPSC line A4. Directed differentiation toward the cardiomyocyte lineage resulted in the production of cells exhibiting ventricular or atrial gene expression profiles, as well as putative trabecular myocardium and potential pacemaker-like cells.
Moreover, the hierarchical clustering of single-cell gene expression revealed a pattern of cardiac, trabecular, and pacemaker segregation that recapitulates biological developmental and expression patterns (Christoffels and Moorman 2009; Christoffels et al., 2000; King et al., 2002; Lyons et al., 1990; Small and Krieg, 2004; Zammit et al., 2000).Together, these results support the pluripotent potential of the reprogrammed cell lines generated in this context and their capacity of differentiation into subspecialized cell types.
Our work describes a previously untested approach to generate pluripotent, cytogenetically normal iPSCs from primary mouse tissue. Reprogramming murine cells with human factors provides a unique opportunity, not only to study the exogenous versus endogenous gene expression of reprogramming factors, but also the kinetics of the virus in reprogrammed cell cultures. In addition, our results highlight the tractability of human OSKM to reprogram divergent species. The safety of Sendai vector systems is of particular relevance with the growing interest in the use of iPSC technology for clinical purposes. These results demonstrate that mouse material provides a reliable test bench for virus-based reprogramming that may be directly translatable to human cells in the clinical setting.
Footnotes
Acknowledgments
We thank Julie Koeman and Dr. Galen Hostetter from the Van Andel Research Institute Cytogenetics and Pathology Core for their assistance with cytogenetic analysis and teratoma tissue identification, respectively. We would also like to thank members of the Jovinge group for thoughtful discussions and feedback, with a special thanks to Laura Winkler and Dr. Eric Kort. An early version of this article was included in the thesis of L.T. This work was supported by generous funds from the Richard and Helen DeVos Foundation.
Authors' Contributions
L.T. contributed to conception and design, collection and assembly of data, data analysis and interpretation, and article writing. E.E. and L.K. provided assistance with collection and assembly of data. S.J. was involved in all stages of the project and provided financial support. All authors provided final approval of article before submission.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.