
Abstract
Abstract
DNA methylation is an essential epigenetic mark for embryo development and can be susceptible to environment factors such as in vitro conditions. The aim of this study was to verify the effect of in vitro culture until Day (D) 14 of the development on the embryo size and DNA methylation pattern of the insulin-like growth factor 2 (IGF2)-imprinted gene. To achieve this, we produced bovine embryos completely in vivo, completely in vitro, and in vitro until D7 and then in vivo up to D14. The embryos produced in in vitro were smaller than those in other two groups (p = 0.024); no differences in embryo size were observed between genders. The in vitro embryos showed a higher level of DNA methylation in the IGF2 as compared with that in the completely in vivo-produced (IVV) embryos (p = 0.009). Furthermore, totally in vitro-produced male embryos showed higher levels of DNA methylation as compared with those observed for the totally IVV male embryos (p = 0.034). No differences were observed among genders for IGF2 DNA methylation. These results showed that the window between D7 and D14 is critical for embryo development and alterations in the environmental conditions during this period can impair DNA methylation establishment of important developmental imprinted genes. This study brings unprecedented data for bovine embryos regarding the impact of the environmental conditions during the posthatching development.
Introduction
DNA methylation is one of the most studied epigenetic events. It occurs preferentially in the regions rich in CG dinucleotides, which can be located in gene promoters of germline differentially methylated regions or in imprinting control regions (Ohlsson et al., 2001; Reik and Walter, 2001). In mammals, DNA methylation regulates many biological processes, such as genomic imprinting, X chromosome inactivation, and transposon silencing (Faulk and Dolinoy, 2014; Gehring et al., 2009; Jeon et al., 2012; Pramio et al., 2016). Moreover, DNA methylation plays a crucial role in gametogenesis and embryo development (Hemberger et al., 2009; Kota and Feil, 2010; Nonchev and Tsanev, 1990; Reik et al., 2001; Sasaki and Matsui, 2008), which justifies the interest in studying these processes in the context of assisted reproductive techniques (ARTs). The effects of ARTs on epigenetic patterns have been evaluated in different studies, such as in in vitro oocyte maturation (Fagundes et al., 2011; Mendonca Ados et al., 2015), sperm sexing (Carvalho et al., 2012), and somatic cell nuclear transfer placenta tissues (Silveira et al., 2018).
Genomic imprinting is a process in which only one of the alleles is expressed depending on the parental origin of the allele (Gebert et al., 2006). One of such imprinted genes is the insulin-like growth factor 2 (IGF2). It is located on chromosome 29 of bovine genome and has a paternal expression (Feil et al., 1994). This gene is involved in embryo development and placentation (Curchoe et al., 2005; Frost and Moore, 2010; Perecin et al., 2009). It is well known that H19 gene shares the same locus with IGF2 and their expression levels are controlled by three intragenic differentially methylated regions (DMRs) and one intergenic DMR (Feil et al., 1994; Moore et al., 1997). Many previous studies have associated alterations in IGF2 methylation patterns with altered imprinting establishment that induces abnormal development in the mammals produced by ARTs (Peters, 2014; Sakian et al., 2015; Yang et al., 2007).
One important characteristic of the epigenetic events is their susceptibility to the environmental factors. Diseases, nutrition, and toxin exposure as well as in vitro conditions can alter DNA methylation patterns, especially in crucial stages of the development, such as gametogenesis and initial embryo development, in which epigenetic reprogramming occurs (Faulk and Dolinoy, 2014). In vitro culture conditions used in ARTs can modify the expression patterns of genes related to cell-to-cell gap junctions, cellular apoptosis, oxidative stress, and cellular differentiation, which can directly affect the embryo development (Rizos et al., 2002) and could be a cause behind the low rates of in vitro embryo production (Diogenes et al., 2017; Lonergan and Fair, 2008). As an example, one of the most controversial elements that are added in culture medium is the fetal calf serum (FCS). Although it is a source of proteins, its utilization is associated with some fetal and placental abnormalities that cause abnormal fetal growth and consequently dystocic parturition (Hoshi, 2003; Wrenzycki et al., 1999; Young et al., 1998).
Most previous studies have evaluated the embryos until Day (D) 7 of the development. However, the morphology of the spherical blastocysts is not a reliable indicator of the quality and ability of the embryos to develop and initiate the pregnancy (Mamo et al., 2011; Pontes et al., 2011). In addition, ∼40% of the embryos, degenerate between D8 and D17 of pregnancy, until maternal recognition (Mamo et al., 2011). In this period of embryo elongation many cellular differentiations, such as the formation of epiblast and hypoblast from inner cell mass, occur (Vejlsted et al., 2006). During this period, around D14 of the development, embryos need to signal for pregnancy recognition and placenta formation, which starts at D18 (Wathes and Wooding, 1980). Hence, this is the time during which the greatest number of pregnancy loss occurs.
Therefore, the posthatching development allows us to evaluate the quality of embryos produced by ARTs at a more advanced stage of development and can provide important information about the elongation phase of those embryos. Earlier studies have shown that in vitro embryos are different from their in vivo counterparts in various aspects, including morphology, metabolism, and gene expression (Diskin et al., 2006; Hansen et al., 2010; Urrego et al., 2017). However, little is known about the effect of the culture system on embryo development and the methylation patterns of embryos. Considering that epigenetic characteristics are susceptible to environmental influences such as culture conditions and the fact that an aberrantly imprinted pattern can cause abnormal development, patterns of IGF2 DNA methylation in posthatching embryos may provide important information about the influences of in vitro culture until D14, on the epigenetic reprogramming.
To the best of our knowledge, no previous studies have evaluated the methylation status of the D14 bovine embryos. Therefore, the aim of this study was to investigate the effect of the in vitro conditions on the embryo culture until D14 on the embryo development and to analyze the DNA methylation of the IGF2-imprinted gene in trophoblast cells.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise specified.
In vitro embryo production up to D7
Ovaries from crossbred cows (Bos taurus indicus × Bos taurus taurus) were collected immediately after slaughter and transported to laboratory in saline solution (0.9% NaCl) supplemented with antibiotics penicillin G (100 IU/mL) and streptomycin sulfate (100 μg/mL) at 35°C. Cumulus/oocyte complexes (COCs) were aspirated from 3 to 8 mm diameter follicles and selected using a stereomicroscope. Only COCs with homogenous cytoplasm and at least three layers of cumulus cells were used. The selected COCs were washed and transferred (groups of 25–30) to a 200 μL drop of maturation medium under silicone oil and incubated for 22 hours at 39°C in 5% CO2 in air. The maturation medium consisted of TCM-199 (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Invitrogen), 0.01 IU/mL of follicle-stimulating hormone, 0.1 mg/mL of
Following maturation, COCs were transferred to a fertilization medium, which consisted of Tyrode's albumin lactate pyruvate (Parrish et al., 1995) supplemented with penicilamine (2 mM), hypotaurine (1 mM), epinephrine (250 mM), and heparin (10 μg/mL). For in vitro fertilization, the oocytes were fertilized with frozen semen from a bull of proven in vitro fertility, which has been used as a reference bull in our laboratory. Motile spermatozoa were obtained using the Percoll (GE Healthcare, Piscataway, NJ) gradient method (90%:45%) in microfuge tubes (Machado et al., 2009) and added into the fertilization drop at a final concentration of 1 × 106 spermatozoa/mL. Spermatozoa and oocytes were coincubated for 20 hours at 39°C with 5% CO2 in air; the day of in vitro insemination has been considered as D0. After the coincubation, presumptive zygotes were transferred to 200 μL drop of SOFaaci culture medium (Holm et al., 1998) supplemented with 2.77 mM of myoinositol and 5% of FCS, and were cultured at 39°C with 5% CO2 in air for 7 days.
Embryos were evaluated for cleavage on D2 postinsemination and for blastocyst rates on D6 and D7 postinsemination. D7 embryos were classified according to the International Embryo Transfer Society (IETS) Manual (Robertson and Nelson, 1998) and only the quality grade-I blastocysts were used for further analysis. Some of the D7 blastocysts (n = 10) were transferred nonsurgically 7 days after estrus, to the uterine horn ipsilateral to the corpus luteum of previously synchronized recipients. The remainder of the embryos were placed in posthatching development (PHD) culture system as previously described by Machado et al. (2013a, 2013b).
In vivo embryo production up to D7
For in vivo-produced (IVV) embryos, crossbred donor females were subjected to superovulatory protocols as described by Machado et al. (2013), and artificially inseminated with frozen/thawed semen from the same bull used for in vitro fertilization, 12 and 24 hours after the ovulation induction. Seven days after artificial insemination, embryos were recovered using double-uterine flushing described previously (Neto et al., 2005). Embryos were evaluated and classified according to the IETS Manual (Robertson and Nelson, 1998) and transferred nonsurgically 7 days after estrus, to the uterine horn ipsilateral to the corpus luteum of previously synchronized recipients.
IVV and in vitro-produced embryo transfer and recovery
Synchronization of the recipients used for transfer of the in vitro-produced (IVP) and IVV embryos was performed according to a previously described protocol (Machado et al., 2013b). The embryos were nonsurgically transferred to the uterine horn ipsilateral to the corpus luteum of synchronized recipients 7 days after estrus. Seven days after the embryo transfer, IVV and IVP D14 embryos, were collected by flushing the uterus twice with 500 mL of phosphate-buffered saline (PBS). The horns of the uterus were carefully manipulated and the embryos were captured in an Erlenmeyer flask. Recovered embryos were isolated in individual drops on a Petri dish, measured using Motic Image Plus 2.0 program (Motic China Group Co. Ltd., Xiamen, China), and morphologically evaluated by observing the parameters of the trophoblast and recording the presence of the embryonic disk. After identification of the embryonic disk, two biopsy samples of the trophoblast tissue were collected with a scalpel blade. One biopsy sample was stored at −80°C in PBS without calcium and magnesium for DNA methylation analysis and the other biopsy sample was frozen at −80°C for sex determination.
In vitro PHD and embryo recovery
The PHD system consisted of tunnels produced in agarose gel covered with culture medium, as described previously (Brandao et al., 2004; Machado et al., 2013b). Only Grade-I embryos were selected for the PHD system, for reasons described elsewhere (Brandao et al., 2004; Machado et al., 2013b). These tunnels were made by dissolving low-melting-point agarose (Gibco BRL, Gaithersburg, MD) in Milli-Q water to a final concentration of 2.4%. A final concentration of 10% FCS was added after the agarose was dissolved. These tunnels were covered with embryo culture medium and used for culturing D11 IVP embryos, as described by Brandao et al. (2004). From D7 to D11, embryos were kept in culture in modified synthetic oviductal fluid (SOF) media as described by Machado et al. (2013b). On D11, only those embryos with a minimum diameter of 0.5 mm, a clear trophoblast, and a compact inner cell mass, were transferred to agarose gel tunnels. The loaded embryos remained in the PHD culture until D14 at 39°C with 5% CO2 in air.
The embryos were morphologically evaluated on D14. Those embryos which increased in size, showed no extrusion cells, no dark spots in the trophoblast, and a well-defined embryonic disk were considered to have a normal appearance. Both, the length of the embryos and the embryonic disk, were measured using the Motic Images Plus 2.0 program (Motic, British Columbia, Canada), in which the device was coupled to a Stereo Microscope. To remove the elongated embryos, one cut was made with a scalpel and blade on the gel and then embryos were slowly withdrawn from the tunnel. The IVV and IVP group embryos were biopsied and two samples of trophoblast tissue were collected with a scalpel blade. One biopsy sample was stored at −80°C in PBS without calcium and magnesium for DNA methylation analysis and the other biopsy sample was frozen at −80°C for sex determination.
Embryo sexing
Embryo sexing was performed by using a multiplex polymerase chain reaction (PCR) with two pairs of primers. The first pair was specific to a region on the Y chromosome, whereas the second pair was specific to a bovine autosomal gene (Table 1). Genomic DNA from known male and female animals were used as positive controls, whereas PCR performed without DNA template was considered as negative control. For PCR, a mix of 20 ηM of each primer pair, 200 μM of each dNTP, 1 × PCR buffer, and 1 U Platinum Taq DNA Polymerase (Invitrogen) was prepared in a final volume of 30 μL. The PCR program details are as follows: 40 cycles of 94°C for 20 seconds, 57°C for 30 seconds, and 72°C for 30 seconds, followed by a final extension step at 72°C for 3 minutes. Amplicons were electrophoresed on a 2.0% agarose gel, stained with ethidium bromide (10 mg/mL), and visualized under ultraviolet illumination. Two amplicons indicated a male embryo, whereas a single amplicon indicated a female embryo. After sex identification, trophoblast biopsies from individual embryos were stored at −80°C for DNA methylation analysis.
Primer Sequences for Embryo Sexing
Bondioli et al. (1989).
Ellis et al. (1988).
DNA isolation and sodium bisulfite treatment
DNA from trophoblast biopsies was isolated using the QIAmp DNA Micro Kit (Qiagen, Hilden, Germany). DNA samples were treated with sodium bisulfite using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA) according to the manufacturer's protocol, except that the conversion temperature was changed to 55°C. Bisulfite DNA-treated samples were diluted with 12 μL of distilled water and stored at −80°C until PCR amplification.
PCR amplification, cloning, and bisulfite sequencing
Sodium bisulfite-treated DNA samples were subjected to nested PCR to amplify a DMR located in the exon 10 of the IGF2 gene (GenBank accession number X53553.1). The molecular structure of the bovine IGF2 gene showing the CpG island that was analyzed in this study is illustrated in Figure 1. The primer sequences, CpG island position, and amplicon size are listed in Table 2.

Representation of the cattle IGF2 gene. Numbered rectangles represent exons, while the square represents the H19 gene. DMR and ICR are represented by small rectangles. The CpG island analyzed in this study is located in IGF2 exon 10. White circles represent each individual CpG that was analyzed. The enhancer (E) that is involved in controlling the H19 and IGF2 genes is represented by two circles. DMR, differentially methylated region; ICR, imprinting control region; IGF2, insulin-like growth factor 2. Color images are available online.
Gene Identification, Primer Sequences, Annealing Positions, and Amplicon Size
Gebert et al. (2006).
IGF2, insulin-like growth factor 2.
Two rounds of PCR amplification were performed in a total volume of 20 μL, using 1 × Taq buffer (Invitrogen), 2.0 mM MgCl2, 0.4 mM dNTPs, 1 U Platinum® Taq polymerase (Invitrogen), 1 μM of each primer (forward and reverse), and 3 μL of bisulfite-treated DNA for the first round and 0.5 μL of the amplicon for the second round of amplification. Both rounds of nested PCR were carried out with an initial denaturing step at 94°C for 3 minutes, followed by 45 cycles (denaturing, annealing, and extension) of 94°C for 40 seconds, 45°C (out) or 40°C (inner) for 1 minute, and 72°C for 1 minute with a final extension step at 72°C for 15 minutes.
After the nested PCR, amplicons were purified by agarose gel electrophoresis using the Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI), according to the manufacturer's protocol. The purified amplicons were then cloned into the TOPO TA Cloning® vector (Invitrogen) and transferred into DH5α cells using the heat shock protocol. Plasmid DNA was isolated using plasmid mini-prep protocol (Sambrook, 2001) and individual clones were sequenced using the dideoxy methodology. Only those sequences with a minimum of 95% of homology and 95% of cytosine conversion were used for further analysis. The sequencing quality was analyzed using Chromas® and the methylation pattern was analyzed by using the BiQ Analyzer® program (Bock et al., 2005). DNA sequences were compared with GenBank X53553.1 accession number for IGF2.
Statistical analysis
The methylation pattern data were compared among experimental groups using ANOVA and Tukey's test or the Kruskal–Wallis and Mann–Whitney tests for analyzing the normality distribution. Pearson correlation index between methylation levels and embryo size was also determined. All the analyses were performed using Systat, version 10.2 (Inc., Richmond, CA) and the results are presented as mean ± standard error of the mean. p-Value of <0.05 was considered significant.
Results
Sex and embryo development
Sex and size of D14 embryos produced completely in vivo (VIVO VIVO), completely in vitro (VITRO VITRO), or in vitro until D7 and then in vivo up to D14 (VITRO VIVO) are presented in Table 3. There were no differences in the size between female and male embryos within each group (Table 3). Moreover, comparing male and female embryos, independent of groups, there were no differences on embryo size (p = 0.598). However, when embryo size was analyzed among groups, VITRO VIVO embryos (20.86 ± 9.86) did not differ from VIVO VIVO embryos (18.67 ± 6.27; p = 0.534), but both were found to be larger than the VITRO VITRO embryos (2.31 ± 0.06; p = 0.024 and p = 0.027, respectively).
Sex and Size of Day 14 Bovine Embryos Produced Completely In Vivo (VIVO VIVO), Completely In Vitro (VITRO VITRO), or In Vitro Until D7 and Then In Vivo Up To D14 (VITRO VIVO)
p
DNA methylation
IGF2 DNA methylation levels were compared among the three groups of embryos (Figs. 2–4). Embryos produced completely in in vitro conditions presented a higher level of methylation than those produced completely in vivo (p = 0.009). The VITRO VIVO embryos did not differ from VIVO VIVO or VITRO VITRO embryos (p = 0.364 and p = 0.075, respectively; Fig. 2).

DNA methylation pattern in the last exon of the IGF2 gene in trophoblast cells of D14 bovine embryos. It was analyzed for embryos produced completely in vivo (VIVO VIVO), in vitro until D7, and then in vivo up to D14 (VITRO VIVO), and completely in vitro (VITRO VITRO). Each line represents individual clone and each circle represents one CpG dinucleotide (28 CpGs in total). White circles represent unmethylated CpGs, filled black circles represent methylated CpGs, and gray circles represent a CpG that could not be analyzed. The numbers at the bottom of each group represent the DNA methylation percentage. DNA methylation percentage of the last exon of the IGF2 gene in VIVO VIVO, VITRO VIVO, and VITRO VITRO bovine embryos is also presented as bar graph. Different letters on bars indicate different DNA methylation patterns between groups within the same sex. Data are represented as mean ± standard error. D, Day.

DNA methylation pattern in the last exon of the IGF2 gene in trophoblast cells of D14 bovine embryos produced completely in vivo (VIVO VIVO; n = 8), completely in vitro (VITRO VITRO; n = 4), and in vitro until D7 and then in vivo up to D14 (VITRO VIVO; n = 10). Each line represents individual clone and each circle represents one CpG dinucleotide (28 CpGs). White circles represent unmethylated CpGs, filled black circles represent methylated CpGs, and gray circles represent a CpG that could not be analyzed. The numbers at the bottom of each group represent the DNA methylation percentage.
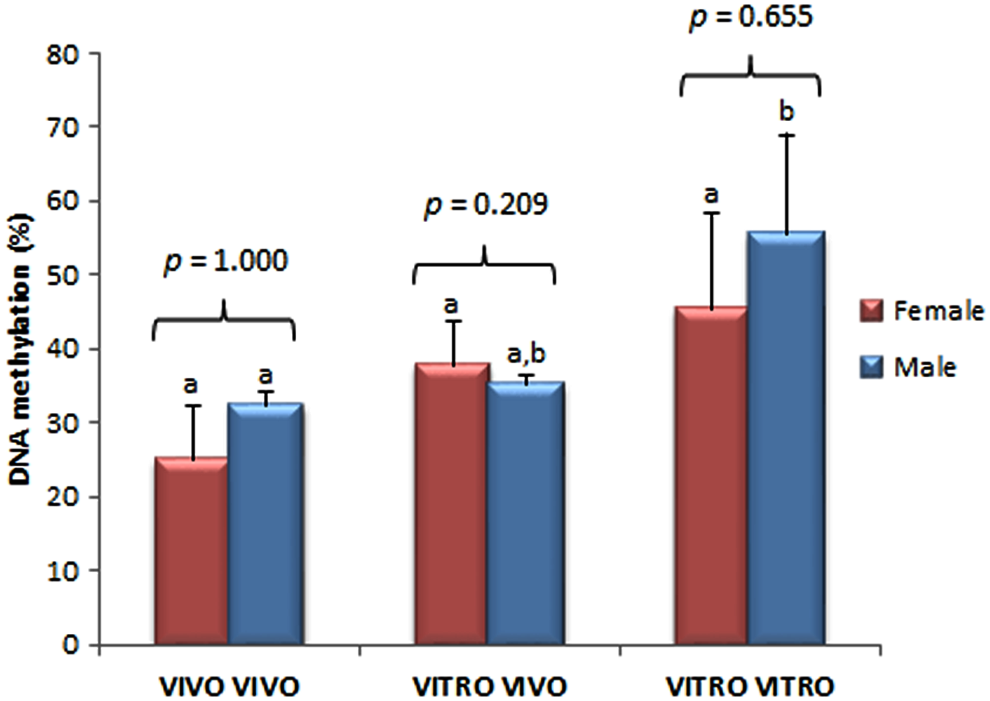
Percentage of DNA methylation of the last exon of the IGF2 gene in trophoblast cells in female and male D14 bovine embryos produced completely in vivo (VIVO VIVO), completely in vitro (VITRO VITRO), and in vitro until D7 and then in vivo up to D14 (VITRO VIVO). Different letters on bars indicates different DNA methylation patterns between the groups but within the same sex. Data are represented as mean ± standard error. Statistical analysis between sexes within the same group is shown as p-values. Color images are available online.
There were no differences observed in DNA methylation levels between female and male embryos within each group (VIVO VIVO p = 1.00; VITRO VIVO p = 0.20; VITRO VITRO p = 0.65). When embryos belonging to only one sex were analyzed, no difference were found among female embryos (p = 0.17). However, such differences were observed for male embryos (p = 0.049), with VITRO VITRO male embryos showing a higher methylation level than the VIVO VIVO embryos (p = 0.034; Figs. 3 and 4).
No correlation was observed between the DNA methylation levels and embryo size (Fig. 5, r = −0.259, p = 0.24).
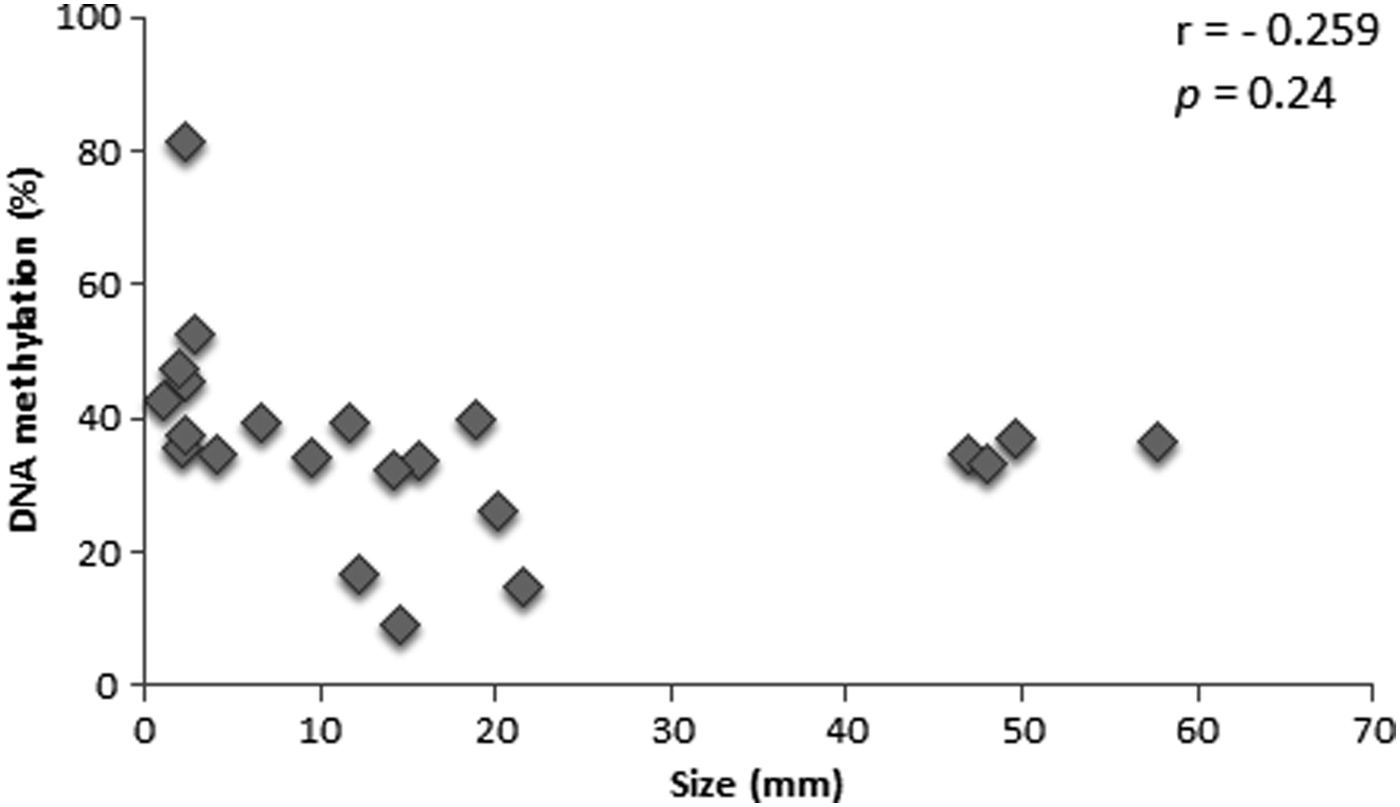
Dispersion plot of percentage of DNA methylation on the last exon of the IGF2 gene in trophoblast cells and embryo size at D14 of development. Each dot represents an embryo with respective size and its profile of IGF2 methylation.
Discussion
It is well established that the in vitro culture can cause such changes during the development that have consequences for fetal growth and postnatal life (Siqueira et al., 2017). In fact, several studies have shown that assisted reproductive techniques (ARTs) affect DNA methylation patterns, genomic imprinting status, and gene expression in mouse, pig, and cow embryos among other species (Canovas et al., 2017; Salilew-Wondim et al., 2015; Wright et al., 2011). Bovine embryos are generally assessed at D7 before transfer, thus the majority of research has focused on the gene expression or developmental potential of blastocysts at this time. Indeed, only a few studies to date, have focused on the elongating conceptus, with the majority emphasizing on the gene expression (Barnwell et al., 2016; Bertolini et al., 2002; Clemente et al., 2009; Machado et al., 2013a; Rodriguez-Alvarez et al., 2010) and none has evaluated the epigenetic patterns.
Considering that placenta is more sensitive to epigenetic perturbations than the fetus (de Waal et al., 2015) and in vitro conditions have been shown to affect the methylation status of several imprinted genes, including IGF2 (Salilew-Wondim et al., 2015), we chose to evaluate the changes in DNA methylation of the trophoblast of D14 embryos produced in different culture conditions. To address our objective, we compared the size and DNA methylation of IGF2 gene in embryos produced in vivo until D14 (VIVO VIVO), embryos produced in vitro up to D7, and then transfer to the recipient's uterus and kept until D14 (VITRO VIVO), and embryos produced completely in vitro until D14 using the PHD system (VITRO VITRO). Since the PHD system allows the evaluation of the developmental potential of IVP embryos beyond D7 and D8, it has emerged as a tool to evaluate the quality of embryos produced by different ARTs at a more advanced stage of development. Our results indicated that complete in vitro culture had a negative effect on the embryo development until D14, which is in agreement with other studies (Brandao et al., 2004; Machado et al., 2012, 2013a, 2013b). However, when embryos were produced in vitro and later were submitted to the posthatching culture in the uterus of a recipient, we noted that embryo growth was restored.
Although no statistical difference was detected in the size between male and female embryos within the group, male embryos were found to be almost twice as large as female embryos in the VITRO VIVO and VIVO VIVO groups. Moreover, the VITRO VITRO embryos were the smallest ones and no difference in size between genders was observed. It is possible that the capacity of the in vitro embryos to protect themselves against the adverse conditions of the PHD environment made the differences between sex disappear. Even though the size of the embryos was similar to that observed in other studies that used the PHD system (Brandao et al., 2004; Machado et al., 2012, 2013a, 2013b), lesser number of female embryos developed in complete in vitro conditions. The effect of the in vitro environment favoring male embryo development has also been reported by those authors.
As far as the IGF2 DNA methylation status was concerned, the VITRO VIVO embryos did not differ from VIVO VIVO, suggesting that uterine environment is particularly important for embryo survival and development in the preimplantation stage (Clemente et al., 2009; Machado et al., 2013a). This hypothesis is based on the results reported by Gebert et al. (2009) who found that on D7 of the development, the methylation of IGF2 was altered as compared with their in vivo counterparts. In our results, no differences in methylation was found between in VIVO VIVO and in VITRO VIVO at D14. Thus, we can suppose that the effect of the in vitro culture on the methylation of IGF2 gene observed on D7 may be minimized by the uterine environment, which contains secreted factors that are important for the survival and growth of the conceptus.
In contrast, we found that the analyzed region of bovine IGF2 was more methylated in embryos cultured in a completely in vitro system than the ones cultured in a completely in vivo system. These data are unprecedented for this embryo stage, and we found that the in vitro culture done until D14, alter IGF2 methylation, at least in D14 embryos. The reason for the altered methylation status in VITRO VITRO can either be the highly adverse environment or the culture time; it has been shown that the longer the embryo is exposed to in vitro conditions, the more changes are observed in the level of methylation (Salilew-Wondim et al., 2015). The changes are visible in both directions: hyper and hypomethylation, indicating a more complex response than a simple delay in either demethylation or remethylation (Sirard, 2017).
We can furthermore suggest that the DNA methylation profile could act as an environmental sensor of the in vitro culture modifications and other stressful conditions. The inferior quality of these embryos can be confirmed by their lower developmental performance. In fact, previous studies from our group have showed that embryos cultured until D14 presented altered characteristics, such as embryo size and gene expression, due to culture conditions (Machado et al., 2013a, 2013b); this may be due to an aberrant DNA methylation profile in VITRO VITRO embryos. In fact, as demonstrated by de Waal et al. (2015), the placenta is more sensitive to epigenetic perturbations than the fetus, and as the number of manipulations increases, the morphological and molecular phenotype of the placenta becomes more severe. The period between D7 and D14 in bovine embryo seems to be essential for the establishment of epigenetic markers such as IGF2 DNA methylation. Alteration in this epigenetic marker can affect embryo development and placenta formation (Curchoe et al., 2005; Frost and Moore, 2010; Perecin et al., 2009).
We also tested the hypothesis that IGF2 DNA methylation pattern in D14 embryos could be different according to embryo sex in vitro. Although no difference in IGF2 DNA methylation was found between genders in each culture condition analyzed, male embryos produced in completely in vitro culture presented higher levels of DNA methylation than male embryos produced completely in vivo.
The literature is contradictory about relation of the embryo sex with culture system. Some studies show that bovine female embryos seem to be more susceptible to environmental conditions, as female blastocyst presented a slower development (Avery et al., 1992) with higher number of apoptotic cells (Ghys et al., 2016) when comparing to their male siblings. Other studies, however, elucidate that male embryos are more sensitive to suboptimal conditions of culture in mouse (Perez-Crespo et al., 2005), humans (Hansen et al., 1999), as well as cattle (Heras et al., 2016). Given that only one VITRO VITRO female embryo was produced, the statistical difference related to sex may have been masked by the low number of samples.
Considering the well-demonstrated relationship between quality and size of the embryos (Lazzari et al., 2002; Mansouri-Attia et al., 2009), we evaluated the relationship between methylation and embryo size. However, we found no correlation between these two parameters.
In conclusion, this study confirmed the potential application of the PHD system as we obtained VITRO VITRO embryos. However, the difficulty to produce female embryos and smaller size of embryos produced in this system underlines the necessity of the improvement of PHD system to be used as a model in future studies. Besides, we were able to detect epigenetic differences in trophoblast cells during the D7–D14 window of development, opening new perspectives of evaluating the relationship of epigenetic changes and placentation. This study brings unprecedented data for bovine embryos regarding the impact of the environment during posthatching period on DNA methylation pattern of an important developmental imprinted gene.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was financed by Embrapa Genetic Resources and Biotechnology, Brazil; the Coordination for the Improvement of Higher Education Personnel (CAPES), Brazil; and Brazilian National Council of Research (CNPq), Brazil.