
Abstract
Abstract
Generation of induced pluripotent stem cells (iPSCs) has been described as a powerful method to dedifferentiate the specialized cells to pluripotency. However, obtaining cancer-specific iPS cells (iPCs) encounters several barriers. The generation of iPCs provides valuable experimental platforms to mimic oncogenesis and offers potentials regarding drug screening. To overcome the difficulties regarding the iPC generation, we aimed at optimizing the generation of iPCs from glioblastoma multiform (GBM) cell lines and at understanding the potential barriers ahead of this process. The T731, T653, and mouse embryonic fibroblast cells were transduced by using retroviral plasmids encoding Oct4, Sox2, and Klf4. The cells were cultured on a layer of feeder cells for 14 days in iPS media and the obtained colonies were then picked and expanded to be evaluated for pluripotency markers by alkaline phosphatase staining, qRT-PCR, and Western blotting. Our findings confirmed resistance in cancer cells to achieve the pluripotency markers. In addition to designing technical tricks to obviate the barriers ahead of iPC generation, we suggested the small molecule PD98059 to enhance the efficiency of iPC generation from GBM cell lines. The resulting iPCs can further be used as a platform to study the mechanism of cancer formation and as a tool for drug screening for the treatment of patients with GBM.
Introduction
Oncogenic transformation is the process through which a normal cell becomes cancerous. Tumorigenesis is a multistep process that is usually initiated by alteration of a single oncogene in normal primary cells, although it is not sufficient to acquire a transformed phenotype in vitro. In many cases, on the oncogene activation or suppression of a tumor suppressor gene, a number of phenotypic changes occur to complete the transformation process. It appears that at least two co-operating oncogenes, which are complementary in their functions, are required. One should induce immortalization of cells, whereas the other completes the oncogenic process by transforming the cells so that they acquire a complete oncogenic phenotype (Land et al., 1983).
Since the discovery of in vitro iPSC generation, the number of cell types that have been reprogrammed has increased exponentially, as this type of iPS cells provides a valuable tool for the study of many diseases, allows drug screening, and has great potential for advances in regenerative medicine (Chao and Chern, 2018). Further, the fact that the cells of origin can be derived from the patient's own cells eliminates the risk of graft rejection.
On the other hand, it does not require the use of biological material from human embryos, thereby avoiding the ethical dilemmas associated with embryonic stem cell (ESC) studies. Possessing these characteristics, tumor cell reprogramming could provide a proper tool for the study of the mechanisms underlying the process of oncogenic transformation (Wu et al., 2017).
However, the number of publications showing successful reprogramming of tumor cells is very low (Ramos-Mejia et al., 2012). The iPC cells are much more difficult to obtain compared with normal iPS cell generation and this seems to be due to the unique genetic and epigenetic pattern of cancer cells (Câmara et al., 2016; Hiew et al., 2018; Ji et al., 2015; Vatanmakanian et al., 2019). To help overcome this difficulty in iPC generation, we have tried to set up an optimized method to generate iPC from in vitro transformed cell lines representing aggressive forms of brain tumor-GBM and to understand the potential barriers ahead of efficient generation of iPCs.
The process of iPC generation largely resembles that of iPS cell generation. Briefly, the process comprises three major steps: introducing exogenous-defined transcription factors into cancer cells, transferring the cells into a medium specific for ESCs, and picking iPC colonies to the passage and examining pluripotency (Sun and Liu, 2011). A remarkable advantage of the iPC models over animal models is that iPCs can be derived from human cells and are even patient derived, thus providing proper research tools to study the molecular mechanisms of oncogenesis; and drug screening.
Like another asset, the reprogramming of cancer cells to pluripotency gives us the opportunity to redifferentiate them and thereby alter their susceptibility to cancer treatment drugs (Kumano et al., 2012; Raya et al., 2009). Although considerable progress has been achieved in the field of iPS and iPC, the researchers are still in a rudimentary phase of disclosing the mechanisms of induced pluripotency and understanding its value in disease recapitulation and clinical applications.
Based on the similarities between the processes of cell reprogramming and oncogenic transformation, as well as scarcity of successful results in generating iPS cells from tumor cells, we plan to study the possibility of iPSC generation from two mouse-derived astrocytes lines transformed by oncogenic events H-RasV 12 (T653 cells) and cRb−/−/RasV 12 (T731 cells) to recognize the obstacles ahead of the iPC generation and try to design optimized methods to efficiently generate iPCs based on the potential technical barriers. Using the two distinct cell lines can also provide a platform in which the characteristics of cancer cell reprogramming can be compared between the cells harboring H-RasV 12 oncogenic events and those combined with a loss of tumor suppressor Rb.
Alternatively, the resulting iPCs have the potential to be a proper platform for further investigations of mechanisms underlying the GBM tumor formation, screening novel drugs, studying the chromatin resetting, differentiation studies, and, ultimately, the capability of iPCs as cell-based therapies.
Materials and Methods
Cell culture
T731, T653, mouse embryonic fibroblasts (MEFs), and Human Embryonic Kidney 293T (HEK293T) cells were maintained in culture in fibroblast medium Dulbecco's modified Eagle's medium (DMEM) (Sigma), supplemented with 10% fetal bovine serum (FBS) (Sigma), 1% penicillin/streptomycin (Sigma), and 1% glutamine (Sigma). The cells were incubated at 37°C with 5% CO2 in air and were passaged every 3 days.
During the reprogramming process, T731 and T653 cell lines were cultured over a layer of feeder cells. ESCs and induced pluripotent stem cells (iPSCs) were also maintained on layers of feeder cells with iPS/ES medium: DMEM (Sigma) or KnockOut DMEM (KO-DMEM; Life Technologies) supplemented with 15% Knockout Serum Replacement (KSR; Life Technologies), 1% penicillin/streptomycin (Sigma), 1% glutamine (Sigma), nonessential amino acids 1X (Sigma), 2-Mercaptoethanol 0.1 mM (Life Technologies), and Leukemia Inhibitory Factor (LIF, 1000 U/mL; Millipore). The cells were maintained in a 37°C incubator with an atmosphere of 5% CO2 and passaged every 3 days.
MEF feeder cell preparation
In passage 3, the MEFs were mitotically inactivated by using mitomycin-C (Inibsa Laboratories) treatment with a final concentration of 10 mg/mL for 2–3 hours. After treatment, the cells were washed extensively with PBS, then trypsinized, and finally counted for freezing aliquots of 3 × 106 cells in freezing medium.
In this study, the feeders were used as a base structure for culturing the cells during the reprogramming process as well as expansion of the obtained iPSCs. To be used, the feeder cells were thawed and cultured on gelatinized plates (gelatin 0.1% w/v in water) in fibroblast medium through the day before transferring the target cells on top of them.
Retroviral infection
To generate retroviral vectors, the HEK293T cells were transfected with the plasmids of interest and pCL-Eco retroviral packaging vector (Addgene plasmid #12371) (Naviaux et al., 1996).
HEK293T cells were plated the day before the transfection at a density of 5 × 106 cells per 100 mm-plate or 2 × 106 cells per 60 mm-plates. For the formation of retroviral particles expressing OSK genes, each of the plasmids pMXs-Oct3/4 (Addgene plasmid #13366), pMXs-Sox2 (Addgene plasmid #13367), pMXs-Klf4 (Addgene plasmid #13370) (all generated in the laboratory of Dr. Yamanaka) (Takahashi and Yamanaka, 2006), and pBabe-puro IRES-EGFP (Addgene plasmid #14430) (generated in the laboratory of Dr. L Miguel Martins, the MRC Toxicology Unit, Leicester, United Kingdom) were co-transfected with plasmid retroviral packaging vector pCL-Eco in HEK293T cells by using the reagent Polyethylenimine (PEI) (Polyscience; #23966) according to the manufacturer's procedure.
Briefly, plasmid DNAs (1 mg/μL) were added to a final concentration of 5 and 2 mg per P100- and P60-plates, respectively (expression plasmids+ retroviral packager). Six micro-liters of the PEI reagent per gram of plasmid DNA was added and diluted in 500 and 200 μL of serum-free DMEM for P100 and P60 plates, respectively. The mixture was then vortexed and incubated at room temperature for 10 minutes. DNA/PEI mixture was added dropwise over HEK293T cells and maintained in a 37°C incubator with 5% CO2. After 24 hours, the supernatant was replaced by fresh medium and incubated for another 24 hours.
The day before transduction, the targets cells (T731, T653, and MEFs) were plated at a density of 1.2 × 106 cells per 100 mm-plates or 6 × 105 cells per 60 mm-plates. After 24 hours, the conditioned transfected HEK293T supernatants containing retroviral particles were collected and filtered through 0.45 μm PVDF filters (JetBiofil). As a transduction facilitator reagent, Polybrene (Sigma) was added to the supernatant at a final concentration of 8 μg/mL. The supernatants were added to the target cells and incubated for 12 hours at 37°C with 5% of CO2. The HEK293T cells were incubated with fresh medium for the next rounds of transduction. We conducted three rounds of infection with the intervals of 12 hours by using the supernatants from 24, 48, and 72 hours transfected HEK293T cells.
As a visual control for monitoring the efficiency of transfection and transduction, the plasmid pBabe-puro EGFP-IRES was transfected into HEK293T cells and the viral particles were transduced into MEFs in parallel. Giving the fact that during generation of T731 and T653 cells, the GFP had been used as control, we transfected/transduced red fluorescent protein (pLJM10 plasmid, addgene) in parallel for these two cell lines as a control of transfection/transduction efficiency. The cells were checked by a fluorescence microscope using green and red filters to estimate transduction efficiency in MEF and GBM cells, respectively.
Cellular reprogramming
For the reprogramming process, MEFs, T731, and T653 cell lines were plated in fibroblast medium with densities of 1.2 × 106 and 6 × 105 per 100 mm- and 60 mm-plates, respectively. In both reprogramming systems, 12 hours after transduction, the media were changed to fresh media to stop the viral infection. After another 12-hour incubation at 37°C with 5%CO2, splitting of the cells was performed on distinct 60 mm-plates in the presence or absence of feeder cells. The next day, all the media were replaced by iPS medium. The media are refreshed every 48 hours during the reprogramming process (14 days).
Colony picking
To expand and characterize the obtained iPS colonies, the media of reprogramming plates were replaced by fresh iPS media 2–3 hours before colony picking. At the time of picking, the medium was substituted by KO-DMEM. Using an optical microscope and a laminar flow hood, the colonies were picked with a micropipette (P20 yellow tip) in a volume of 10 μL of medium and transferred individually to 0.2-mL Eppendorf tubes, into which we added a further 30 μL trypsin (trypsin-EDTA 1X) (Sigma). The tubes were then incubated in the incubator at 37°C and 5% CO2 for 5 minutes.
Subsequently, 70 μL of iPS medium was added to each tube to neutralize the trypsin function. Next, the suspension was pipetted up and down 10 times to disrupt the colonies. The content of each tube was seeded in a well of a 96-well plate with feeders to a final volume of 100 μL with iPS medium plus two signaling inhibitors PD98059 (3 mM) (Selleckchem) and CHIR99021 (3 Mm) (Selleckchem) to facilitate the formation of iPS colonies. The cells were maintained in an incubator at 37°C and 5% CO2. The medium was replaced daily with iPS medium in the presence of PD and CHIR (Modified protocol of Li et al. 2011).
When the cells reached 80%–90% of confluence, they were trypsinized and expanded to larger plates (usually to 24-, 12-, and 6-well plates, respectively) until 100 mm-plates with feeders from which the population expanded, aliquoted, and froze in iPS freezing medium or were used for confirmative tests.
Alkaline phosphatase staining
Alkaline phosphatase (AP) staining was performed at day 14 of reprogramming. AP activity of iPS cells was detected colorimetrically by using the BCIP (5-bromo-4-chloro-3-indolyl-phosphate)/NBT (nitro blue tetrazolium) Color Development Substrate commercial kit (Promega) according to the manufacturer's instructions. After washing with PBS, the cells were fixed by 4% formaldehyde v/v in PBS and incubated for 1–2 minutes at room temperature. The plates were then treated with staining solution composed of 5 mL of AP buffer (100 mM Tris-HCl [pH 9.0], 150 mM NaCl, 1 mM MgCl2), 33 μL of NBT, and 16.5 μL BCIP and placed in a dark place for 30 minutes. The plates were then washed and maintained in PBS.
Protein extraction
Protein extraction was started by trypsinization of cells in culture. The cells were then washed with PBS and precipitated by centrifugation at 3500 rpm for 5 minutes. The cell pellets were then lysed with double volume of RIPA buffer (150 mM NaCl, 10 mM Tris-HCl pH 7.5, 0.1% SDS, 1% Triton, 5 mM EDTA pH 8.0, 1% deoxycholate) accompanied with inhibitors, 1 mM sodium orthovanadate, 1 mM DTT, 4 mM NaF, 1 mM cocktail of protease inhibitors (Sigma), and 1 mM PMSF. After 20 minutes of incubation on ice, the lysate underwent sonification for 5 minutes and was then centrifuged at 14,000 rpm for 15 minutes at 4°C. The supernatant was transferred to a new tube and then quantified.
To quantify, the Bradford method using the DC Protein Assay (BIO-RAD) commercial kit was performed according to the manufacturer's recommendations. A serial dilution of bovine serum albumin (BSA) (Sigma) was provided for simultaneous absorbance measuring of the samples. The absorbance was measured at 750 nm in a 96-well plate reader VERSAmax microplate reader (Molecular Devices). The sample concentrations were determined by using a standard curve designed based on known BSA concentrations.
RNA extraction
For the isolation of total RNA from cells in culture, the commercial kit RNeasy Mini Kit (Qiagen) was used following the manufacturer's recommendations. RNA quantification was performed in a NanoDrop Spectrophotometer 2000c (Thermo Scientific). Two micrograms of the RNA sample was applied to cDNA synthesis reaction.
Western blotting
Fifteen microliters of each protein extract (with the concentration of 2 μg/μL) was electrophoresed through a 12% polyacrylamide gel (30% solution Acrylamide/Bis 29:1; BIO-RAD). Separated bands were then transferred to 0.45 μm PVDF membranes (Millipore). Primary antibodies and horseradish peroxidase-coupled secondary antibodies were used to identify the proteins of interest. To visualize the target bands, a chemiluminescent-enhanced chemiluminiscent-based detection system was used, and finally the treated membranes were exposed to auto-radiographic films for 30 minutes to visualize the expressed proteins.
The primary antibodies used were: anti-OCT 3/4 (rabbit polyclonal; Santa Cruz Biotechnology sc-9081; 1: 500 dilution); anti-SOX2 (polyclonal goat; Santa Cruz Biotechnology sc-17320; 1: 500 dilution), anti-KLF4 (polyclonal rabbit; Santa Cruz Biotechnology sc-20691; 1: 500 dilution), anti p44/42 MAPK (ERK1/2) (monoclonal rabbit; Cell Signal 9102; 1:1000 dilution), anti P-p44/p42 MAPK (ERK1/2) (Thr202/Tyr204) (monoclonal rabbit; Cell Signal 4370; 1:2000 dilution), NANOG (Millipore, AB 5731; 1:5000 dilution), anti-α-ACTIN (mouse monoclonal; ICN; 1:2000 dilution), and anti-β-TUBULIN (mouse monoclonal; Millipore AA2; 1:2000 dilution).
cDNA synthesis and quantitative RT-PCR
To convert RNA to cDNA, High-Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, CA) was used according to the kit instructions. The reaction was performed in the c1000 Thermal Cycler (BIO-RAD). Quantitative PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems) in a thermocycler Stratagene Mx 3005P (Agilent Technologies). The expression levels were presented relative to Gapdh as housekeeping gene in each sample with the ΔCT method (Momeny et al., 2018). The primer pairs used in our study were according to Table 1.
Primer Pair Sequences Used for Quantitative Reverse Transcriptase-Polymerase Chain Reaction Experiments
Endo, endogenous sequence of the genes.
Statistical analysis
For the statistical analysis of the results, we used the Student's t test based on the means and standard deviations of the obtained data. Owing to the small test and control cases, the Mann–Whitney test was used to compare the results and a p-value of <0.05 was considered statistically significant.
Results
Long-term culture of Glioblastoma cells at a high confluence is not viable
To optimize the culture conditions for reprogramming the GBM cells into iPS cells, we initially performed a prolonged culture, avoiding passaging of cells, in 60 mm-plates. It was previously established for MEF cells that 600,000 cells seeded per each 60 mm-plate are well tolerated during the process of reprogramming. Despite this, and probably due to loss of contact inhibition, in the case of GBM cells (T731 and T653), this starting number of cells showed a detrimental effect in the culture, with the cells forming huge masses detaching from the surface of the plates approximately in the first week of the culture. The lower number of cells, such as 200,000 and 100,000 cells, was also not tolerated for this process in which the cells required at least 14 days of continuous culturing.
Finally, an initial number of 50,000 cells per 60 mm-plate for GBM cells was found to be optimal to proceed with the experiments. The initial MEF number of 600,000 cells was used as a positive control of reprogramming. This observation implies that the high proliferation rate of cancer cells might be an obstacle for long-term maintenance of the culture and suggests that lowering the starting number of cancer cells for the reprogramming process might be a good strategy to obviate this barrier facing the successful reprogramming of cancer cells.
MEF feeder cells support GBM cell maintenance during the reprogramming process
Feeder layer cells usually include adherent, growth-arrested but biochemically active and viable cells. They have been used for a number of years to promote cell proliferation, especially for the cells grown at low density. Utilizing such cells that are unable to divide but yet capable of secreting growth factors provides a tool to maintain ESCs as well as iPSCs. As mentioned earlier, we reduced the initial amount of GBM cells used for the reprogramming process. As a result, we reasoned that using feeder cells could be a great help to maintain the cancer cells during the reprogramming process.
We have shown in our experiments that using feeder cells as physical and biochemical support of GBM cells is crucial throughout the long-term culture (Fig. 1A–D). In addition, to prevent detaching of the cells during the process, applying feeders seems to improve the process, since this resulted in the formation of more AP-positive (AP-positive) colonies in reprogrammed GBM cells than their feeder-free counterparts. In our first attempt to reprogram the T731 cell line with separate retroviral OSK, it was demonstrated that despite having the initial number of 50,000 cells grown on the top of feeder cells, the resulting AP-positive colonies were much more than the feeder-free cells with an initial number of 100,000 cells per 60 mm-plate (Fig. 1E).
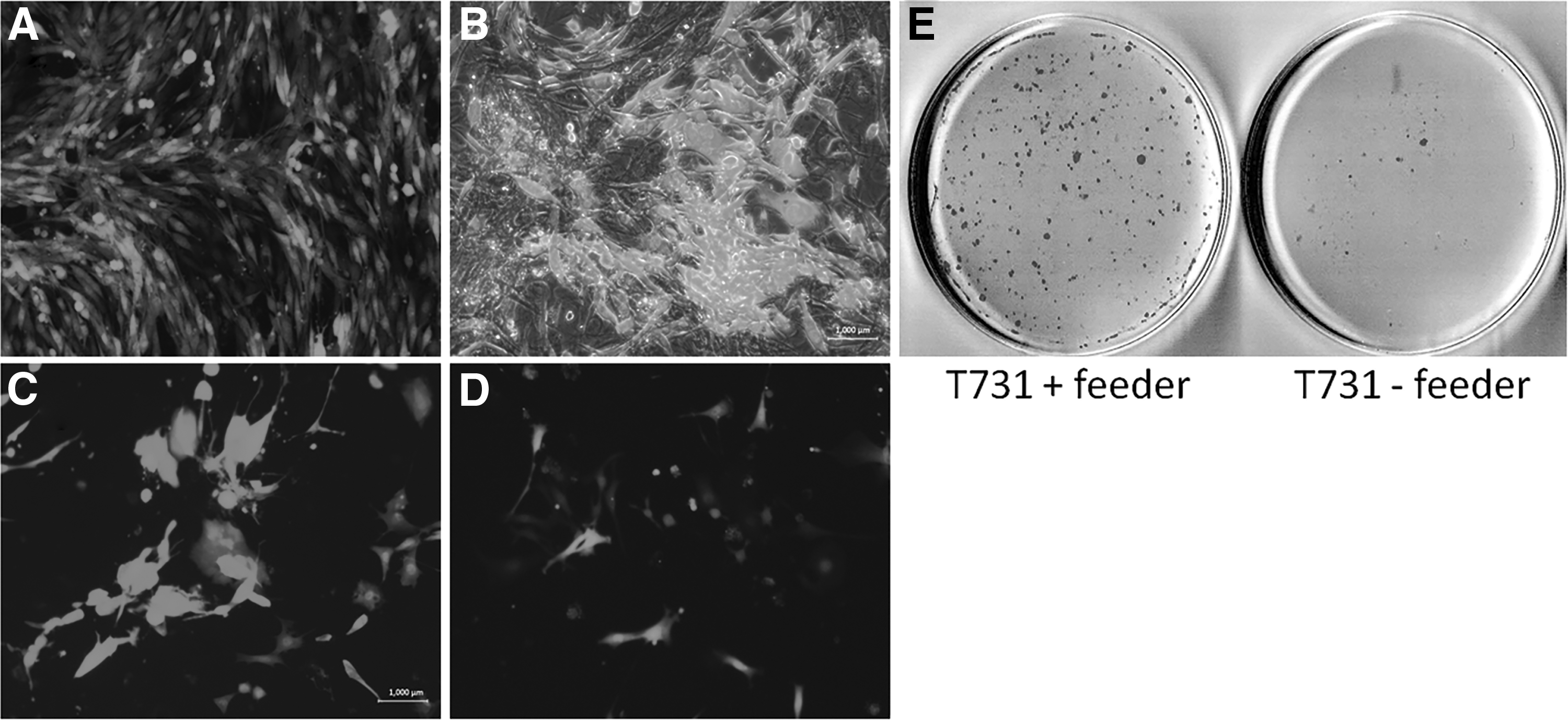
The impact of using MEF feeder cells as a growth supporter of GBM cells during reprogramming. The
These results suggest that incorporating the feeder cells in reprogramming of oncogenically transformed cells not only helps to maintain the target cells during the long-term culture but also has a beneficial effect by enhancing the emergence of pluripotency markers and improving the reprogramming efficiency.
IPS-like colonies obtained from GBM cells are resistant to be expanded
To decipher whether the iPS-like colonies resulting from GBM cells can eventually be expanded to a pure iPS cell population, we designed a new set of an experiment in which we tried to pick the putative iPS-like colonies and expand them serially into multiwell plates seeded with feeder cells.
Further, we launched a new experiment with an equal initial amount of cells (T731, T653, and MEFs) to provide comparable conditions regarding the cell numbers and using feeder cells, to eventually obtain solid results in which the differences between the efficiency of reprogramming in the target cells could be compared. To do so, the target cells were transduced with retroviral OSK and then transferred over a layer of MEF feeder cells with 50,000 cells per each 60 mm-plate. The RNA and protein extracts from all the target cells were shown to be highly enriched in mRNA and protein levels of exogenous Oct4, Sox2, and Klf4 at day 3 of reprogramming (Fig. 2), confirming the highly efficient transduction of the three reprogramming factors.

mRNA and protein expression of the exogenous OSK at day 3 of reprogramming. Quantitative real-time PCR analysis confirmed significant increases in mRNA expression levels of Oct4
The plates were analyzed regarding the expression of AP at day 14 of reprogramming. The obtained AP-positive spots were analyzed and counted with an equally significant threshold by using ImageJ software version 1.0. Our results demonstrated that, despite having higher colony counts in the GBM cells (269 and 265 colonies for T731 and T653 cells, respectively) compared with the MEFs (114 colonies) (Fig. 3A), the GBM cells failed to form typical iPS colonies, and the AP-positive colonies appeared as a disseminated positive population lacking the defined borders of a real iPS population (Fig. 3B). Thus, the higher colony counting in GBM cells cannot be attributed to higher efficiency of generating iPS cells in GBM cells over MEFs, since the resulting colonies are not able to show real morphological and molecular characteristics of iPS cells.
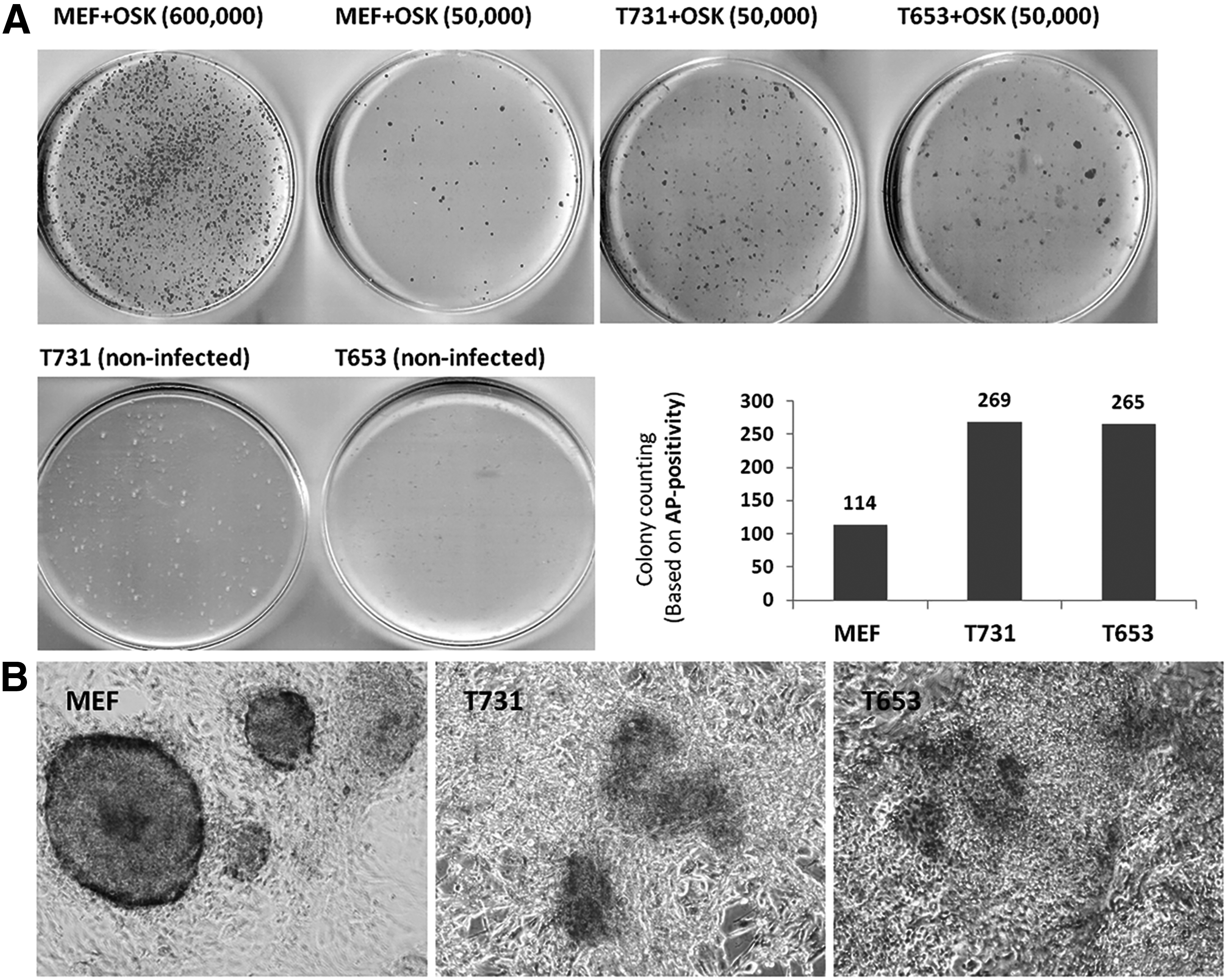
AP staining and colony counting in target cells transduced with OSK. The
The suspected colonies were then picked and passaged into 96-well plates to track whether they can be purely expanded or their ability to express pluripotency markers. Based on previous studies (Ying et al., 2008) to support colony formation and to maintain the cells in an undifferentiated state, we used a small-molecule inhibitor of glycogen synthase kinase 3 (Gsk3) CHIR99021, in combination with the inhibitor of mitogen-activated protein kinase kinase (MEK) PD98059. The single colonies picked from MEF cells were efficiently expanded in a 96-well plate (12 of 12 picked colonies were grown with typical colonies). The MEF-derived iPS colonies were also passaged and expanded through 24,- 12,- and 6-well plates and subsequently expanded in 60 mm- and 100 mm-plates (Fig. 4).

Expansion of picked iPS-like colonies derived from MEF, T731, and T653 cells. The figure indicates the well-expandable colonies derived from MEF cells through different passage stages, forming numerous typical and pure iPS colonies. Although T731 cells were resistant to form colonies, T653 cells were inclined to create several iPS-like colonies through the expansion period. It seems that the developed iPSCs from GBM cells tend to suppress GFP, and the suspected iPS-like colonies in T653 cells appear as black holes under fluorescent microscopy (white arrows).
On the other hand, the colonies picked from T731 cells were not forming typical colonies during expansion. The cells were grown as small round-shaped cells resembling iPS cells, which also included some differentiated-like areas (Fig. 4). Interestingly, 4 out of 12 suspected colonies picked from putative reprogrammed T653 cells generated colonies that resembled iPS cells during the expansion period. This would suggest that the T653 cell line, carrying fewer oncogenic events, may be more susceptible to the iPS-like formation.
PD98059 treatment increased the number of AP-positive colonies in GBM cells
Owing to the presence of an overactive form of the RAS signaling pathway (H-RASV12) in the T731 and T653 cells, we wondered whether blocking this pathway could enhance colony formation in these cells. To achieve this, we chemically blocked MEK, a key member of the RAS/MAPK pathway, by using a small-molecule inhibitor of MEK, PD98059.
In addition, to find out the optimal interval for PD98059 treatment, we exposed the cells to the inhibitor during days −3 to 0, as well as days 0–3 of reprogramming. On day 3 of reprogramming, protein extract from GBM cells (PD-treated and nontreated cells) was analyzed by Western blotting for the presence of phosphorylated ERK (P-ERK), a phosphorylated substrate downstream of MEK, to track the inhibitor effect. The Western blot analysis confirmed MEK inhibition as judged by the very weak or nonvisible band corresponding to P-ERK in the PD-treated GBM cells compared with nontreated cells (Fig. 5B). Also, the cells seemed to change their morphology during the process (Fig. 5A). To monitor the influence of PD treatment on reprogramming efficiency, we performed AP staining at day 14 of reprogramming.

The effect of PD treatment on morphological changes, P-ERK protein expression, and AP-positive colony formation. The
Interestingly, the results demonstrated a significant increase in AP-positive colonies in the cells treated with PD98059 during either the days −3 to 0 or the days 0–3 of reprogramming (Fig. 5C). The GBM cells did not show almost any positive colony (average 0–2 AP-positive colonies) in nontreated cells with an initial amount of 25,000 cells seeded per well of the six-well plate, whereas the PD-treated cells (treated during both days −3 to 0 and days 0–3) showed an average count of 7 positive colonies. We speculate that the Ras pathway blockage at the initial time points of reprogramming may be an effective strategy to promote iPSC colony formation from GBM cells.
Expanded cells from T653-derived iPS colonies express pluripotency markers
To explore whether the cells expanded from suspected GBM-derived iPS colonies have changed their identity toward pluripotency, we analyzed the mRNA levels of several genes that are representative of pluripotency. The quantitative real-time PCR results indicated that the mRNA levels of endogenous Oct4, Sox2, and Nanog, which are well-known pluripotency markers, were significantly upregulated in cells expanded from colonies obtained from MEFs and T653 cells compared with the parental cells; whereas T731 showed no significant difference between the expanded cells after reprogramming and the parental counterparts (Fig. 6). The Western blot analysis also confirmed the upregulation of NANOG protein in iPS cells obtained from MEFs and T653 cells.
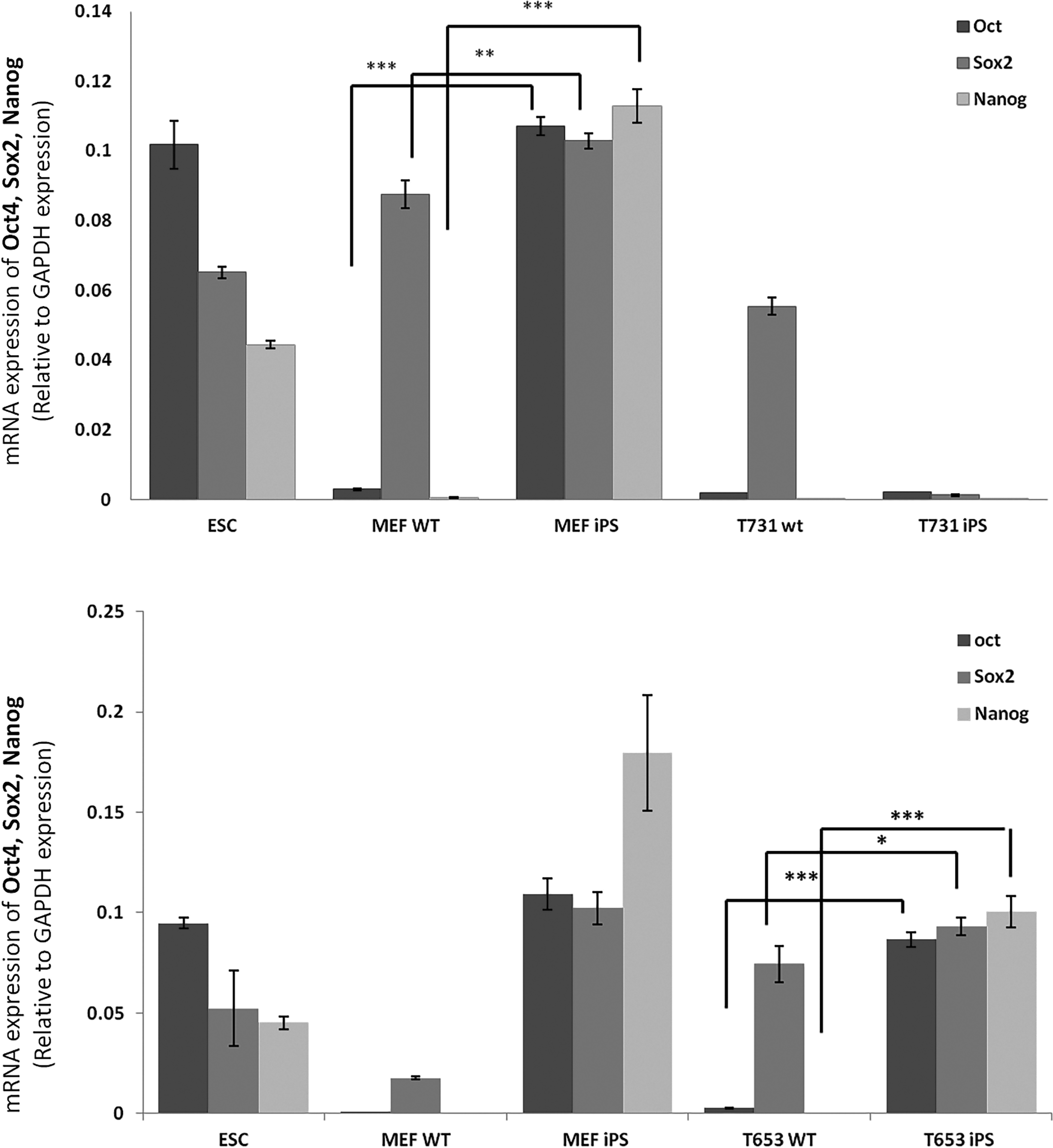
mRNA expression of endogenous Oct4, Sox2, and Nanog, and protein levels of Nanog in MEFs, T731, and T653 cells both before and after reprogramming. The chart indicates a significant increase in the expression of the three pluripotency genes in iPS cells derived from MEFs and T653 cells, whereas the T731 cells did not show enhancement in the markers after the reprogramming process. The expression levels have been normalized to GAPDH expression as a housekeeping gene. Statistical significance was analyzed by Student's t-test. ***p < 0.001, **p < 0.01, *p < 0.05.
These results suggest that despite having morphological changes or irregular expression of AP in colonies derived from T731 cells, they have not been successfully reprogrammed to fully show the minimal requirements of pluripotency. On the other hand, it seems that the T653 cells, which showed more flexible morphologic properties during the reprogramming process, can switch their identity to pluripotency easier than T731 cells that carry more oncogenic events.
Discussion
Reprogramming and oncogenic transformation are stepwise processes that have many similarities in common; thus, iPSCs generated from cancer cells could clarify molecular mechanisms behind the biology of cancers. Unleashing the barriers underlying the reprogramming process of cancer cells could uncover the information on the interconnection between pluripotency and oncogenic transformation that would be a basal platform for therapeutic advancement (Ramos-Mejia et al., 2012).
Alongside, in this study, we attempted the reprogramming of oncogenic cell lines representing GBM, an aggressive form of brain tumor, and the deciphering of the potential barriers ahead of iPC generation. Besides describing several technical adjustments that may help to design experimental procedures for iPC generation, the major achievement of this study was to suggest a chemical inhibitor of the Ras signaling pathway (MEK inhibitor, PD98059) to efficiently enhance iPS colony formation in GBM cells.
Main observations of our study confirmed a highly resistant identity of cancer cells that hinders their efficient conversion to pluripotency. In this research, we have overcome various technical obstacles in the path of GBM cell maintenance during the reprogramming process. We have observed that the characteristics in GBM cells that preclude them to have a normal course of the reprogramming process seem to be diverse and include an abnormal long-term culture, development of atypical and nonorganized AP-positive colonies, resistance to be expanded into a pure iPSC population at least in the cell line with more oncogenic hits (T731), and, finally, impeding the expression of pluripotency markers in amplified suspected colonies in the majority of expanded colonies.
To achieve a better design for cancer cell reprogramming, we first tried to set up a long-term culture adjustment for GBM cells, which is an obligatory requirement of the reprogramming process. Having an aggressive proliferation of these transformed cell lines, it appears that they are not able to tolerate the essential long-term culture with the normal initial amount of cells used for reprogramming of MEF cells (600,000 cells/60 mm-plates). To sort out this problem, we decided to lower the initial amounts to 50,000 cells per 60 mm-Petri dishes or 25,000 cells/well of six-well plates; which were well endured for at least 14 days.
However, growing a very small population of cells in culture requires a strategy to feed the cultured cells regarding their requirements for growth factors and other metabolic needs that are obtained by cell
In addition to having several limitations regarding maintenance of GBM cells throughout the reprogramming process, it appears that their resistance for having an efficient reprogramming is not restricted to these properties. Our observation indicated that the GBM cells have a particular resistance to be purely converted into the iPS cell population. As our AP results showed, the AP-positive colonies derived from the GBM cell source failed to express AP in defined and typical colonies with the pure iPS-like population. Although the AP-positive colonies were shown to be more abundant in GBM cells compared with MEF cells with the same initial amounts, it cannot be interpreted as having a higher reprogramming efficiency, since the quality and capability to be purely expanded were in minimal condition for GBM cells.
Although the AP-positive colonies taken from MEF cells as reprogramming control were observed microscopically to have well-characterized borders resembling ESC morphology, having colonies containing a mix of AP-positive and -negative cells in GBM cells makes the interpretation difficult regarding their pluripotency. To better characterize them in terms of expressing the pluripotency markers, we tried to expand the suspected population over a layer of feeder cells. It seems that the GBM cells harbor another property by which the obtained colonies resist to be expanded as efficiently as colonies derived from MEF cells, and this added another layer of complexity to their resistant characteristics in terms of iPC generation. In the subsequent expansion period, it was shown that a small proportion of picked colonies from T653 cells were successfully expanded and created colonies better resembling iPS-like colonies during the expansion days.
However, the resulting cells expanded from T731 were shown to fail to express the pluripotency markers. On the other hand, we were wondering whether the colonies picked from suspected T653 cells that emerged to have morphologies more similar to iPS-like colonies express pluripotency markers. The qRT-PCR results revealed that the iPCs generated from T653 cells are endogenously enriched in mRNAs of Oct4, Sox2, and Nanog genes that are typical pluripotency markers.
This advantage of T653 over T731, although in a very low efficiency (4 over 12 picked colonies), may be attributed to the fact that T653 cells harbor fewer oncogenic events (having a wild-type form of Rb tumor suppressor while T731 lacks this gene that prevents them from having an oncogenic manner in terms of resistance to iPC generation). This may explain that lacking the Rb tumor suppressor in T731 might be a culprit of the more resistant property of this cell line to reprogramming. We wondered whether blocking this oncogenic pathway can enhance their reprogramming efficiency by diminishing their malignant behavior.
Interestingly, the AP staining results showed significantly greater AP-positive colonies in the GBM cells treated with PD98059 during either the days −3 to 0 or the days 0–3 of the reprogramming process. This observation suggested the overactive RAS/MAPK signaling pathway as a possible underlying mechanism by which the cancer cells are resistant to be efficiently reprogrammed. Moreover, it proposed specific blocking of the cell signaling pathways that are overexpressed in cancers by utilizing the small molecules as a method to promote the iPC generation with greater efficiency.
However, it is highly essential to characterize the iPS colonies obtained from PD-treated cells regarding the pluripotency markers as well as monitoring them in terms of in vitro and in vivo tumorigenicity and the possible disadvantages that required further scrutiny.
Importantly, some small molecules have also been reported to be able to replace some transcription factors in iPSC generation. For example, a G9a inhibitor, BIX01294, was reported to induce iPSCs from neural stem cells, in place of Oct4 (Shi et al., 2008). It was reported that Kenpaullone could substitute Klf4, although the underlying mechanism is still unclear (Lyssiotis et al., 2009). In addition, a transforming growth factor-β inhibitor (616452) could replace Sox2 during iPSC generation (Ichida et al., 2009; Maherali and Hochedlinger, 2009; Woltjen and Stanford, 2009).
Having potential to be used in clinical applications as well as the risks of retroviruses, it would be essential for iPCs to be free of viral infections; thus, discovering novel methodologies replacing the forced expression of reprogramming factors using lentiviral or retroviral agents can be a great benefit in terms of using iPC as therapeutic cellular sources. More studies are needed to shed light on different mechanisms behind the resistance of tumor cells to reprogramming as well as providing new knowledge to technically support the iPC generation.
Conclusions
Taken together, in our study we have described several limitations in two oncogenic transformed cell lines representing GBM cancer, which prevent them from being efficiently reprogrammed to pluripotency. Alongside, we have offered a better design by which the cancer cells can be maintained and reprogrammed through more optimized conditions during the reprogramming process. Our findings highlighted the importance of small molecule PD98059 as an MEK-ERK inhibitor to enhance the reprogramming efficiency of cancer cells to pluripotency. We have also suggested that the cancer cells with higher oncogenic events (T731, harboring H-RasV 12 and lacking Rb tumor suppressor) show a more resistant property regarding the iPC generation compared with the cells with fewer hits (T653 with H-RasV12 alone).
Designing more optimized technical conditions and describing novel strategies to replace the viral agents in reprogramming cocktail can lower the risk of mutagenesis and tumorigenicity of iPCs, which can make them safer to be applied for regenerative medical purposes.
Footnotes
Acknowledgments
The authors would like to declare their special thanks to Mr. Farid Solaymani Mohammadi for his kind help regarding the language editing of this article. This work was granted by the Tehran University of Medical Science, deputy of research, Grant Number: 9211264003; allocated for the Master thesis of Mr. Mousa Vatanmakanian.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.