
Abstract
Complications of central nervous system in type 2 diabetes mellitus (T2DM) often lead to cognitive impairment and seriously affect the quality of life. However, there is no individualized disease model. Urine-derived stem cells can be an ideal source for generating human induced pluripotent stem cells (hiPSCs) and progenitors, as they are easily accessible, noninvasive, and universally available. In our research, we differentiated urine-derived hiPSCs into neuron (N), astrocyte (A), and microvascular endothelial cells (E) from a T2DM patient. Next step, we intend to coculture these three cells together in a 3D system to create a new disease model in vitro, which may simulate the cerebral microenvironment of DM, for future study of its pathogenesis and precision medical treatment.
Introduction
The injures of diabetes mellitus (DM) in the central nervous system (CNS) are mainly characterized by diabetes-induced cognitive impairment, neurophysiological and structural changes of the brain. The complications of DM injury to the CNS include cognitive decline, vascular dementia (VD), depression, epilepsy, and so on. In the past 20 years, the cognitive ability of middle-aged people with diabetes has decreased by 19% compared with those without diabetes (Mayeda et al., 2015). Especially, substantial evidence is accumulating that type 2 diabetes mellitus (T2DM) are related to cognitive impairment, and T2DM is an established risk factor for developing dementia and Alzheimer's disease (AD) (Albai et al., 2019; Biessels et al., 2006; Kopf and Frolich, 2009).
In addition, the risk of AD or VD in patients with T2DM is 1.5–2 times higher than that in normal people (Biessels et al., 2006). However, further studies related to molecular mechanisms of brain injury with DM have not been well elucidated. In our preceding work, we found that the T2DM brain injuries, such as cognitive impairment and hemichorea, had complicated and personalized relationship with the neuron (N), astrocyte (A), microvascular endothelial cell (MEC) (E), and other target cells. Cognitive impairment of DM was mainly related to the decrease of neurons in frontal lobe and hippocampus, proliferation of astrocytes, and destruction of blood-brain barrier, diabetic hemichorea is mainly related to the changes of astrocyte quantity and morphology (Lin et al., 2013; Moghaddam et al., 2014; Prasad et al., 2014; Shan et al., 1998; Wang et al., 2014).
However, the factors and molecular signaling pathways that cause N, A, and E cell injury/activation are still unclear. At present, most studies on diabetic brain injury used animal disease models, which will take a long time and have a low success rate, and there are some differences between biomedical animal models and human disease. There is no individualized model for further study of its pathogenesis.
Stem cells represent a great versatile cell source, as they are able to undergo a very high number of divisions because of their self-renewal property and furthermore differentiate into almost all adult cell types thanks to their pluripotency characteristic (Bordoni et al., 2018). Induced pluripotent stem cells (iPSCs) own similar properties with embryonic stem cells (ESCs) in terms of self-renewal, pluripotency, gene expression, proliferation, morphology, and telomerase activity (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). In the past decades, the human induced pluripotent stem cells (hiPSCs) have found revolutionary application in vitro models of various neurological diseases, such as AD, Parkinson disease, and amyotrophic lateral sclerosis (Dimos et al., 2008; Iovino et al., 2015; Soldner et al., 2009).
In addition, hiPSCs can circumvent the ethical concerns and immune rejection that obstructed the application of human embryonic stem cells (hESCs), and eliminate the shortcomings of species differences and individualized differences in animal models. Its unique potential of self-renewal makes it an exciting candidate for cell replacement therapy for various diseases, such as neurodegenerative diseases and cancers, and offers unlimited possibilities for understanding early disease development and establishing in vitro disease models (Dakhore et al., 2018). Urine specimens is a novel and noninvasive approach to isolate patient-specific stem cells by easy and low-cost procedures and negligibly invasive way and retain up to 0.1%–4% of reprogramming ability that is much more than blood cells and fibroblasts (Falzarano and Ferlini, 2019; Raab et al., 2014).
Furthermore, repeated freeze-thaw cycle will not exert an influence on its reprogramming ability (Raab et al., 2014). Urine-derived stem cells (USCs) are a good source of cells for generating iPSCs and importantly, they can also be directly converted into targeted cell types and opened the door for great potentials to promote regenerative therapies into the clinic (Falzarano and Ferlini, 2019; Sauer et al., 2016; Wang et al., 2016).
Thus, we hypothesized that reprogramming of USCs into N, A, and E may be a new noninvasive strategy for the study of molecular mechanisms of disease pathogenesis, test potential drug toxicology reaction, and develop personalized therapeutic strategies for diabetic brain injury.
Materials and Methods
Ethics statement
All procedures involving animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006), and the study design was approved by the animal Ethics Committee of Dalian Medical University (Dalian, China).
Establishment of hiPSC lines
Urinary cells isolation and expansion
After obtaining written informed consents from the donors, the urine samples were obtained from a 54-year-old male patient with T2DM in the Second Hospital of Dalian Medical University. The patient signed informed consent before urine collection. Three hundred to 500 mL of fresh midstream urine was collected in sterile containers, then centrifuged at 400 rpm for 10 minutes, and washed with 10 mL phosphate-buffered saline (PBS).
After repeating the above steps twice, the cell pellet was resuspended in 0.5 mL of Urineasy full-medium I (cellapybio, China) and maintained in Matrix-coated plates. During the next 3–7 days, half of the medium was replaced with fresh medium every 2 days as follows: suctioned 2 mL medium, centrifuged at 200 g for 5 minutes, the supernatant was discarded leaving 200 μL, added 2 mL Urineasy full-medium II, and then put it back to the plates. Replace the medium every 2 days with 3 mL Urineasy full-medium II after the cell successively attached. After reaching about 90% confluency, cells were passaged in the appropriate ratio (Fig. 1).
Urinary cell isolation and expansion.
Reprogramming of human urine renal epithelial cells
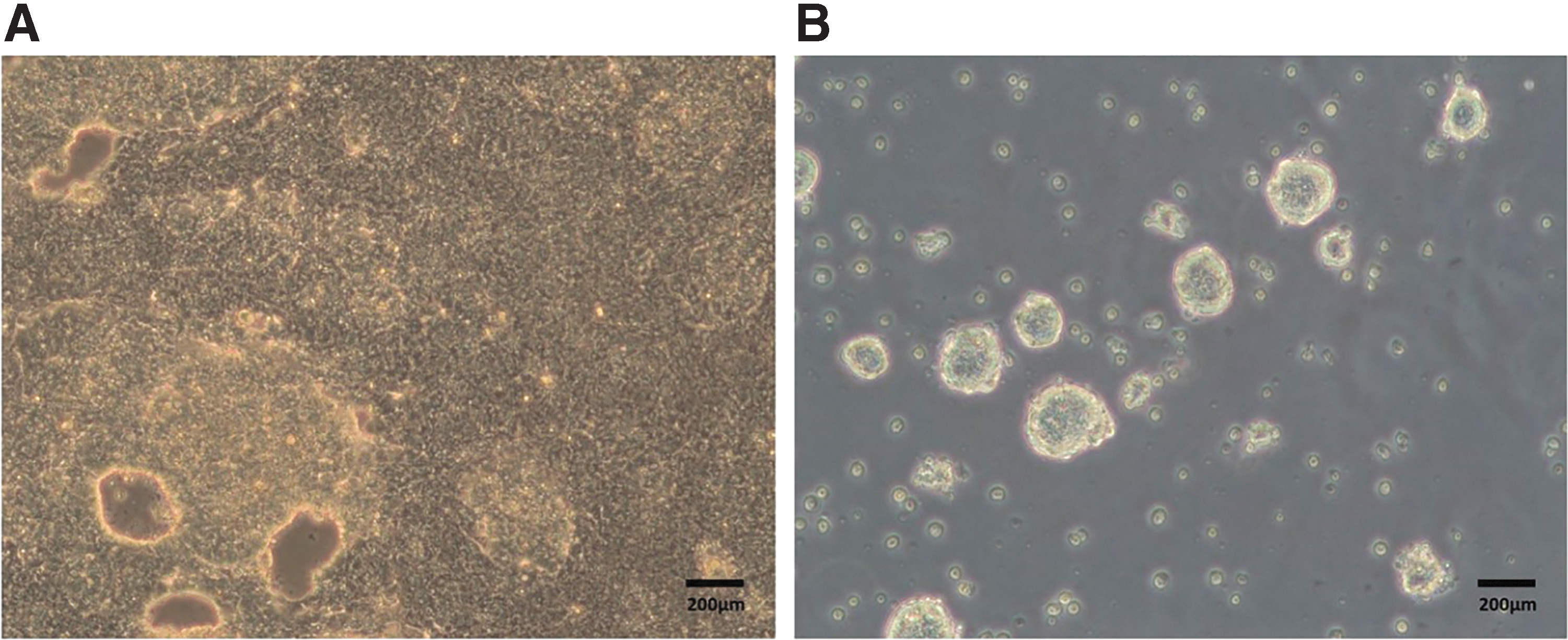
To generate hiPSC lines, the reprogrammed urine cells were cultured in Urineasy full-medium II until reaching confluence of about 80%. According to the manufacturer's instructions, these cells were reprogrammed into pluripotent stem cells by using a nonintegrated iPS 2.0 Sendai Reprogramming Kit—CytoTune™. The CytoTune-iPS 2.0 Kit imported the Sendai RNA virus into the reprogramming factor (SeV, KOS, Klf4, and c-Myc). The medium was changed every 2 days. Inoculated the transfected cells in a culture dish coated with Matrigel containing urine maintenance medium on day 7. The plates were observed for the emergence of characteristic cell colonies every day. Around 3–4 weeks after transfection, picked undifferentiated colonies and transferred them onto fresh Matrigel-coated plates for further expansion and analysis (Fig. 2).

Reprogramming of human urine renal epithelial cells.
Identification of pluripotency of hiPSC lines
Immunofluorescence staining
Immunofluorescence staining was used to analyze the expression of specific multipotent markers. The methods were slightly similar to that of Kyung-Ok Uhm (Uhm et al., 2017) and Zhang et al. (Zhang et al., 2018). Cells were fixed in 4% paraformaldehyde for 30 minutes, permeabilized with 0.3% Triton X-100 (Sigma) for 20 minutes, blocked in 1% bovine serum albumin for 40 minutes, and incubated with primary antibodies (Table 1) for TRA-1-60, TRA-1-81, and SSEA-4. Cell nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI, Vector). Images were acquired using a fluorescence microscope (Carl ZeiSS, Germany) (Fig. 3A).

Antibodies Used for Immunocytochemistry
Alkaline phosphatase staining
BCIP/NBT Alkaline Phosphatase Color Development Kit was used to analyze the expression of specific pluripotency. Cells were incubated with antibodies labeled with alkaline phosphatase, washed 3–5 times with PBS for 3–5 minutes each time, added BCIP/NBT staining solution, incubated in dark for 5–30 minutes, and then washed twice with distilled water. AP stained (red/purple) pluripotent colonies and the differentiated (clear) colonies were observed by a light microscope with 10 × objective (Fig. 3B).
Detection of reprogramming vector
After 10 passages, reprogrammed hiPSC lines were tested for SeV residues. The real-time polymerase chain reaction (RT-PCR) was performed as standard protocol. PCR was performed using primers (Table 2) and instructions as recommended by the manufacturer. As positive control RNA was used from the reprogramming leftovers. Negative control RNA was obtained from the ESC line H9 ( Fig. 3C).
Primer Sequences for Sendai Viral Test
Karyotyping
GTG-band method was used to analyze the karyotype of hiPSCs by standard cytogenetic procedures. Then cells were treated with colcemid (50 ng/mL) for 6–8 hours, incubated in KCL hypotonic solution at 37°C for 20–40 minutes, and fixed with fresh methanol/glacial acetic acid (3:1). The karyotype was detected and analyzed using the VideoTesT-Karyo 3.1 system (Fig. 3D).
Teratoma test
To test the capacity of the reprogrammed hiPSC lines to spontaneously differentiate into cells of all three germ layers, the formation and differentiation of embryoid body were analyzed. We injected 1 × 107 reprogrammed hiPSCs subcutaneously into 4-week-old CB-17 SCID male mice to generate teratomas. The tumors were excised after 2–3 months, fixed in 4% PFA for 24 hours, and embedded in paraffin. The embedded paraffin block was cut into 10 μm, oven at 60°C for 1 hour and baking at 75°C for 2–3 hours. Then hematoxylin and eosin (H&E) staining was done to observe the formation of the three germ layers (Fig. 3E).
Methylation analysis
We used bisulfite sequencing PCR for methylation analysis. Genomic DNA was treated with bisulfite, and then primers were designed on the periphery of CpG island (excluding CpG loci) for PCR. According to gene sequence information, the primers (Table 3) were designed and synthesized by primer5 software. Finally, the PCR products were sequenced. The CPG methylation of OCT 4 and NANOG promoters were determined by real-time quantitative polymerase chain reaction (qRT-PCR) on the GeneAmp 9600 PCR assay system (ABI Bio-Rad, Hercules). PCR cycle was used as follows: 96°C for 2 minutes, followed by 30 cycles of 95°C for 15 seconds, 50°C for 20 seconds, and 60°C for 4 minutes (Fig. 3F).
Primer Sequences for Pluripotency
Directional differentiation of hiPSC lines
Culture hiPSCs
The hiPSCs were cultured in PSC easy human pluripotent stem cell (ESC/iPSCs) culture medium (Cellapy: Cat. no. CA1001500/CA100110 0). The cells were subcultured for differentiation when the confluence reached 80%. We used PSC easy complete medium and cell culture level 0.5 mM EDTA subculture working fluid (Cellapy: Cat. no. CA3001500) to subculture hiPSCs.
Differentiation of iPSCs into neural stem cells
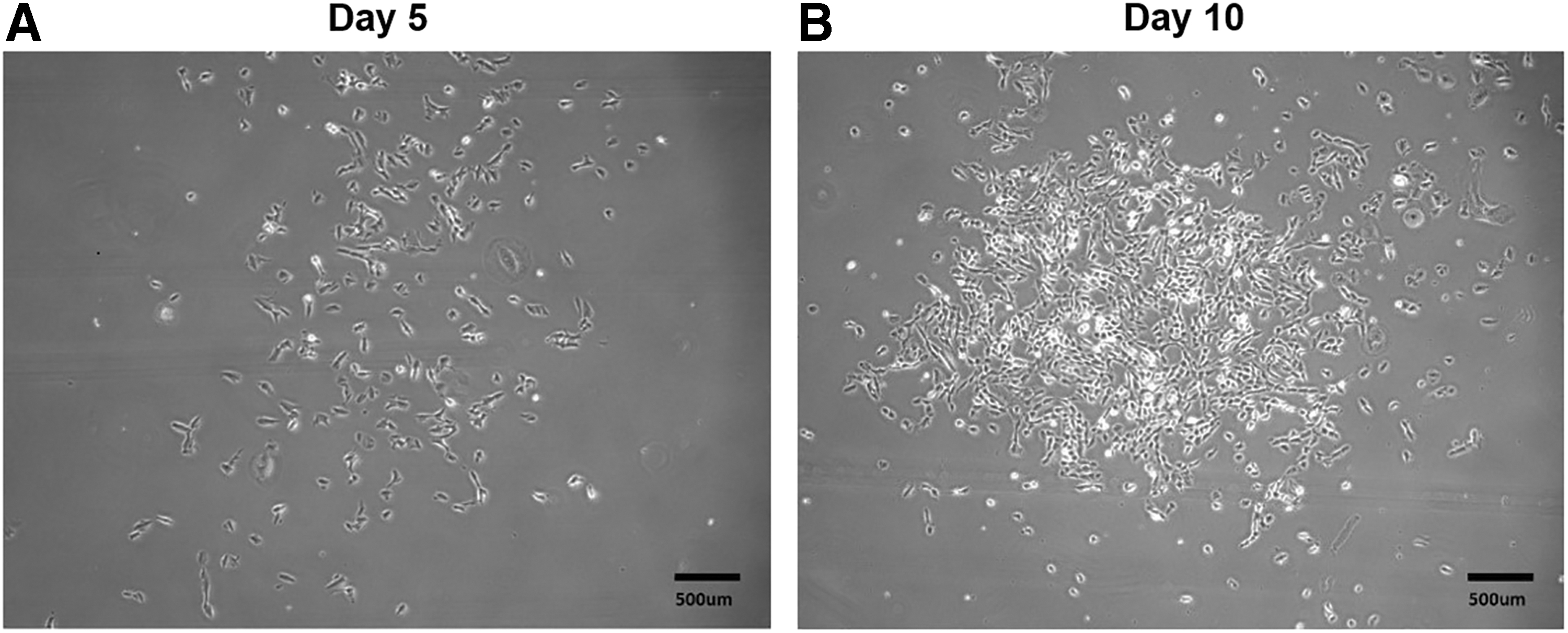
The cells were differentiated when the confluence reached 100%. PSC easy complete medium was removed and washed with 1 mL PBS (Hyclone: Cat. no. SH30256.01). Added 3 mL nerve stem differentiation complete medium to each pore cell, cultured in a 37°C constant temperature incubator with 5% CO2, after 48 hours added 2 mL nerve stem differentiation complete medium again, continuously cultured for 48 hours, and then discarded 2 mL supernatant; the complete culture medium of nerve stem differentiation of 2 mL was added again and cultured in incubator for 48 hours. Microscopically, when Rosette structure appears (Fig. 4A), cells can be digested for suspension culture. The cells were separated by Human Neural Stem Cells (NSCs) Digestible Fluid and resuspended in Neural Trunk Inoculation Medium. The cells were cultured in Human NSC Maintenance Medium for 4–6 days and then subcultured (Fig. 4B).
Differentiation of iPSCs into NSCs.
Differentiation of NSCs into neurons
NSCs, which were subcultured 2–3 times, were collected in a 15 mL tube centrifuging 3 minutes at 800 rpm. Discarded the supernatant, added 1 mL Human Nerve Cell Digestive Juice, digested at 37°C for 15 minutes. Discarded the supernatant after having centrifuged at 1000 g for 5 minutes, then resuspended in Human Nerve Differentiation Inoculation Medium. Next day, replaced the medium with the Human Nerve Differentiation and Maintenance Medium when the cells stuck to wall under microscope. Replaced the medium every 2 days, and neurofilaments could be observed after about 5–9 days (Fig. 5A).

Differentiated cells.
Differentiation of NSCs into astrocyte
The method is similar to differentiation of neurons, slightly different. After the NSCs were digested, the cells were resuspended in Neural Astrocyte Differentiation Medium . Replaced the medium every 2 days, target cells were obtained in about 7 days (Fig. 5B).
Differentiation of hiPSCs into MECs
Digested hiPSCs with EDTA when the confluence reached 80%. Resuspended cells were counted and inoculated into six-well plate according to 3 × 104 cells/cm2. Added 10 μm Y27632 and cultured with PSC easy complete medium. After 48 hours, absorbed the PSC easy complete medium and added the induction medium prepared in advance according to the 2 mL/hole. After 5 days, the medium was replaced by EC+RA medium. MECs were obtained after about two DAPIs (Fig. 5C).
Identification of differentiated cells
Immunofluorescence staining was performed to identify the morphological feature of differentiated cells. Cells were fixed in 4% paraformaldehyde for 30 minutes at 4°C. After washing with PBS, the cells were permeabilized with 0.3% Triton X-100 for 20 minutes, blocked in 1% bovine serum albumin for 40 minutes, and incubated with primary antibodies at room temperature. Cell nuclei were labeled with DAPI (Vector, 20 minutes, room temperature). For detection purposes, different fluorescently labeled secondary antibodies were selected depending on the specifications of the manufacturer. The antibodies are listed in Table 1. Images were acquired using a fluorescence microscope (Carl ZeiSS) (Fig. 6).
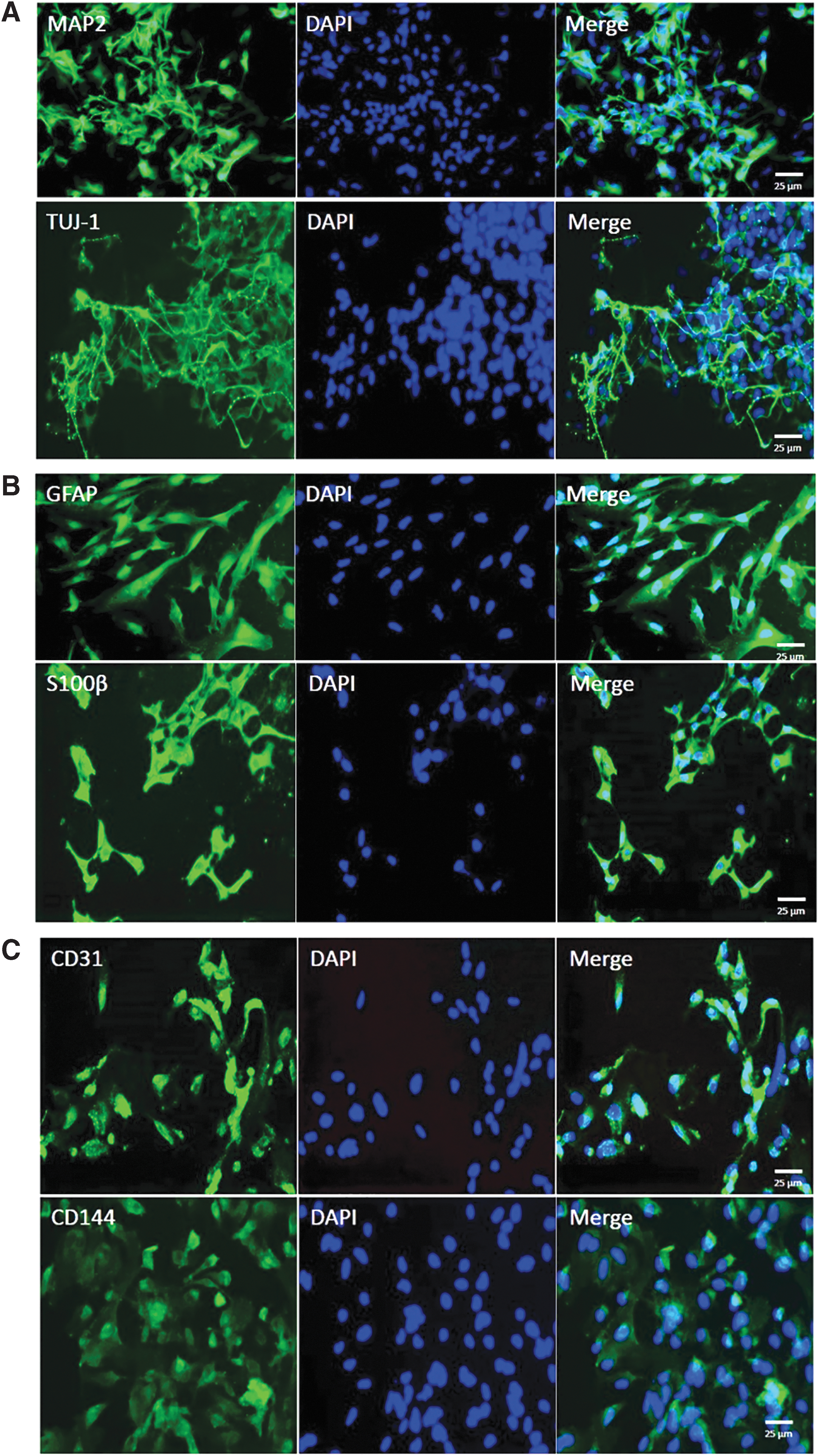
Immunofluorescence staining of differentiated cells.
Results
Characterization of hiPSCs and analysis of pluripotency
We utilized immunofluorescence and AP staining to evaluate the presence of specific pluripotency markers. hiPSCs were observed for the expression of hESC-specific transcription factors NANOG, OCT 4, and SOX2, in addition to cell surface markers SSEA4, TRA-1-60, and TRA-1-81. hiPSC lines showed positive immunofluorescence labeling for above six hESC-specific antigens (Fig. 3A). iPSCs is stable as it can maintain the morphological characteristics of cells and continue to express these specific markers after repeated generations.
Meanwhile, the pluripotency of hiPSCs colonies displayed positive reaction for AP staining (Fig. 3B). Through highly sensitive RT-PCR methods, the Sendai viral sequences were undetectable in reprogrammed iPSCs after 10 passages (Fig. 3C). All hiPSC lines were tested to be normal (46 chromosomes, XY) by GTG-banding karyotypic analysis (Fig. 3D). To further demonstrate the pluripotent status of hiPSCs in vivo, cells were subcutaneously injected CB-17 SCID male mice. All laboratory mice generated teratomas containing representative cell types of all three germ layers, as shown by H&E staining (Fig. 3E).
To identify the epigenetic modification in reprogrammed hiPSCs, we examined the methylation level of CpG dinucleotides in promoter regions of ESC-specific genes OCT 4 and NANOG by bisulfite genomic sequencing (Fig. 3F). In T2DM patient, the CpG methylation ratios in OCT 4 and NANOG promoters of hiPSCs were 2% and 14%, which almost corresponded to 0% and 12% in H9 (ESCs). Moreover, the CpG methylation ratio in OCT 4 promoter regions of somatic cells was 78% in T2DM patients, respectively, significantly higher than that of hiPSCs and H9. Results revealed that the OCT 4 and NANOG promoter regions were demethylated in hiPSCs relative to the somatic cells and were thus similar to those in hESCs. These results further indicated that the reprogrammed hiPSCs preserved ESC-like properties.
Characterization of neuron, astrocyte, and MEC
NSCs derived from hiPSCs can proliferate and differentiate into both neural and glial lineage as stained by specific neuron markers MAP2, TUJ-1 (Fig. 6A), and astrocyte markers glial fibrillary acidic protein (GFAP), S100β (Fig. 6B). MECs differentiated from hiPSCs were stained by CD31 and CD144 (Fig. 6C). Immunocytochemical staining of above three cell types indicated cells stain positive for their specific markers, suggesting high reliability of our study.
Discussion
Currently, a dominant challenge to our comprehension of the pathogenic mechanisms of diabetic brain injury has been the lack of physiologically relevant in vitro models that capture the precise patient genotype and phenotype, with the cell type of interest and specific protein expression. iPSCs technology, together with further cellular differentiation, supplies an attractive methodology for modeling neurological diseases, and for relevant drug discovery. With comparison to mouse counterparts, human iPSCs are considered to not only hold the similar self-renewal and pluripotency capacities of hESCs, but also generate a limitless supply of targeted cell types for in vitro researches.
Our previous studies and a series of clinical and basic researches on DM indicated that diabetic and hyperglycemia metabolic dysfunction led to various degrees of cognitive impairment and damage of learning and memory abilities (Jafari et al., 2009; Lee et al., 2018; Lin et al., 2013; Wang et al., 2014; Zhou et al., 2017). Studies found that there were some pathological changes in streptozotocin-induced diabetic rats/mice, such as neuronal apoptosis, synaptic alterations, increased expression of astrocyte GFAP, and reduced expression levels of glial cell line-derived neurotrophic factor, which will potently promote the survival of many types of neurons (Baydas et al., 2003; Cai et al., 2018; Sadeghi et al., 2016).
Moreover, diabetes and continuous high blood glucose could directly induce a vast spectrum of injuries of endothelial cells in cerebral vascular system (Xu et al., 2016), damage the permeability of BBB by downregulating the expression of occluding and claudin-5 (Yoo et al., 2016). Therefore, here we successfully created hiPSC-derived neuron (N), astrocyte (A), and MEC (E), which displayed typical morphological characteristics and can be used to reveal the pathogenesis on molecular level and explore the innovative treatment strategies based on drug screening or other methods to target diabetic brain injury.
In 2015, the Obama administration announced the plan for precision medicine initiative by the National Institutes of Health (Collins and Varmus, 2015; McCarthy, 2015). Precision medicine is defined as an approach to disease treatment and prevention targeted to maximize effectiveness on the basis of individual characteristics of genes, phenotype, environment, and lifestyle. As we know, DM results from a complex physiologic process that is determined by a large number of risk susceptibility genes and compounded environmental factors. Personalized model of brain injury with T2DM based on patient-specific iPSCs can avoid the genetic background issue and predict drug response of an individual, which help advance the field of precision medicine.
With the precision medicine initiative, it is clear that the generation of patient-derived iPSCs technology will be possible, which can reserve a patient's genetic and molecular background, and will pave the way toward precision medicine goals of utilizing individual data for diagnosis and therapy.
Next work, we will add the sample size and improve the experiment and unravel diabetic encephalopathy pathogenesis by utilizing these differential disease-relevant cell types (N, A, and E). Coculturing these three cells together in a 3D system, combined with advanced microfluidic chip technology, to further create a brand new personalized human brain cell models in vitro, which will provide a new platform to further explore the cellular target spot and molecular mechanism of the type 2 diabetic brain injuries identification, intervention, and evaluation, so as to reveal and establish new direction, strategy, and method on effective diagnosis and treatment, in addition to specific marker and medical target spot of diabetic brain injuries. Therefore it can establish precious and strong scientific foundation and technical support for relevant innovative drug research.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was supported by funding from the National Natural Sciences Foundation of China (NSFC 81571237) and Liaoning Science and Technology Planning Project (2017225070).