
Abstract
Prostate cancer stem cells (PrCSCs) are responsible for the development of castration-resistant disease and are associated with poor outcomes; however, the origin of PrCSCs is still not known due to the lack of a suitable model. In the current study, the human prostate cancer cell line 22RV1 was used to generate induced pluripotent stem cells (iPSCs) via the exogenous expression of four classic transcription factors (OCT-4, SOX2, KLF4, and C-MYC). The iPSCs were analyzed by phase contrast microscopy, real-time polymerase chain reaction, immunofluorescence, alkaline phosphatase (AP) activity, and examined for karyotype and embryoid body and teratoma formation. The analyses demonstrated that the prostate cancer cells were successfully reprogrammed into iPSCs by characteristic human embryonic stem cell morphology, cell marker expression, AP activity, embryoid body, and pluripotency capability in generating all three embryonic germ layers. These results may provide a convenient and accessible model for studying the origin of PrCSCs and the process by which progenitor cells are transformed into PrCSCs.
Introduction
Prostate cancer is one of the most common types of cancer, and the second leading cause of cancer death in the United States (Siegel et al., 2019). Although chemotherapy and androgen deprivation therapy can induce partial or almost complete cancer regression, the regression is typically temporary in patients with advanced disease, and recurrent disease can be resistant to further treatment.
Recently, it has been proposed that prostate cancer cells arise from a small population of cells, termed prostate cancer stem cells (PrCSCs), which possess the potential for self-renewal potential and multipotential properties that allow them to form tumors (Pardal et al., 2003). Although the numbers of PrCSCs are low, study has suggested that they may be responsible for the development of castration-resistant disease and poor outcomes because of their self-renewal potential and because of their common exhibit of drug-resistance (Rybak et al., 2015).
However, the origin of PrCSCs is unknown. A number of studies have suggested that cancer stem cells may be derived from normal tissue stem cells because of characteristics such as self-renewal and the potential for unlimited replication (Lathia et al., 2020; Nimmakayala et al., 2019). Because stem cell activity is tightly controlled, determination of the mechanisms by which normal stem cells are transformed into malignant stem cells may lead to the development of novel anticancer therapies.
Oncogenic mutations are believed to be important in the process of malignant transformation, and one method of studying the interactions of oncogenic mutations is to reprogram an established cancer cell line into pluripotent stem cells. Reprograming differentiated cells into stem cell-like cells may allow the generation of sufficient numbers of stem-like cancer cells for study. This is important because the isolation of stem cell-like cancer cells from human tumors is very challenging due to their very low numbers.
Yamanaka et al. recently reported that somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by transient ectopic overexpression of the transcription factors OCT-4, SOX2, KLF4, and C-MYC (Takahashi and Yamanaka, 2006; Wernig et al., 2007). This discovery provides a potential method for reprogramming cancer cells into iPSCs. Other groups have succeeded in reprogramming cancer cells using different sets of transcription factors (Utikal et al., 2009). However, attempts to reprogram prostate cancer cell lines have not been successful (Borges et al., 2015).
In this study, we generated iPSCs from the human prostate cancer cell line 22RV1 (22RV1-iPSCs) via an integration-free system. To the best of our knowledge, this is the first report of the successful generation of iPSCs from prostate cancer cells. This method may assist in understanding the carcinogenesis of prostate cancer and provide a valuable method for disease modeling.
Materials and Methods
Cell line and culture
The prostate cancer cell line 22RV1 was supplied from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and the cells had been tested and authenticated. Cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (Gibco), penicillin (100 U/mL), and streptomycin (100 mg/mL) (Life Technologies). Cells were grown in a humidified incubator at 37°C with 5% CO2. Early-passage cells were used for the experiments.
Generation and culture of human prostate cancer cell-derived iPSCs
The 22RV1 prostate cancer cells were seeded onto six-well plates and grown to 50%–80% confluency. The transduction assay was performed using the CytoTune®-iPS 2.0 Sendai Reprogramming Kit (Catalog number A16517; Invitrogen Life Technologies), following the manufacturer's protocol. On day 1, the transduction medium was replaced with standard prostate cancer cell culture medium, and the medium was replaced every 48 hours. On day 6, the infected cells were seeded onto Matrigel (BD) precoated plates in mTeSR1 medium (STEMCELL), and the mTeSR1 medium was changed daily during generation. The iPS-like colonies were selected at day 16–20, and cultured in mTeSR1 on Matrigel. The culture medium was changed daily, and the culture conditions were 37°C in a humidified incubator containing 5% CO2.
Staining for alkaline phosphatase
Alkaline phosphatase (AP) staining was done as previously described (Qin et al., 2008). In brief, the culture medium was gently aspirated and cells were rinsed with phosphate-buffered saline (PBS). The cells were then fixed with 4% paraformaldehyde at room temperature for 2 minutes and rinsed twice with tris-buffered saline Tween-20. Freshly prepared AP staining solution (50 mg/mL nitroblue tetrazolium with 50 mg/mL 5-bromo-4-chloro-3-indolyl phosphate) was added in plates and incubated in the dark at room temperature for 15 minutes. The staining solution was aspirated, and plates were rinsed with PBS.
Nonintegrating analysis by reverse transcription-polymerase chain reaction
Total RNA was isolated from cells using TRIzol (Invitrogen Life Technologies). First-strand cDNA was synthesized using a SuperScript® VILO cDNA Kit according to the manufacturer's instructions (Invitrogen Life Technologies). cDNA was amplified by polymerase chain reaction (PCR) using ExTaq (Takara), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the positive control. The PCR primers used for the amplification of each gene are shown in Table 1. Amplified DNA fragments were resolved by electrophoresis on 2% agarose gel and visualized by ultraviolet fluorescence.
List of the Primer Sequences for Polymerase Chain Reaction Studies
AR, androgen receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NANOG, Nanog homeobox.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen Life Technologies) and reverse transcribed with a reverse transcription kit according to the manufacturer's instructions (Takara Biotechnology Co., Ltd., Dalian, China). Quantitative analysis was performed LightCycler® 480 SYBR Green I Master (Roche) on a LightCycler 480 System (Roche), according to the manufacturer's instructions. The PCR conditions were as follows: 95°C for 30 seconds, 35 cycles at 95°C for 5 seconds, then 60°C for 30 seconds. GAPDH was used as an endogenous control. The relative mRNA expression was calculated using the 2−ΔΔCt comparative CT method. Primer sequences are shown in Table 1.
Karyotype analysis
22RV1 and 22RV1-iPSCs were subjected to karyotype analysis, as previously described (Wang et al., 2013). Cells were grown in 10 cm plates, and demecolcine (Dahui Biotech) was added to achieve a final concentration of 50 μg/mL. After 40 minutes, the cells were then trypsinized, pelleted by centrifugation at 200g for 5 minutes, resuspended in 8 mL of 0.075 M KCl, and incubated for 20 minutes at 37°C. A fixative solution composed of one part acetic acid and three parts methanol was added to a final volume of 10 mL, the suspension was mixed gently and incubated for 10 minutes at 37°C.
After further centrifugation, the supernatant was removed and an ice-cold fixative solution composed of one part acetic acid and three parts methanol was added to achieve a final volume of 10 mL. The cells were dropped onto a cold slide and incubated at 75°C for 3 hours, and then were treated with trypsin and colorant, and metaphase states were analyzed using an Olympus BX51 microscope.
Immunofluorescence assay
Cells were fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.1% Triton X-100 in PBS (Sigma-Aldrich) for 10 minutes, blocked in 4% normal goat serum (Cayman Chemical) for 30 minutes, and incubated with primary antibodies in 4% normal goat serum overnight at 4°C in the dark. The next day, the cells were washed three times with PBS and incubated with secondary antibodies (1:200) for 1 hour at room temperature in the dark. After washing three times with PBS, the cells were incubated with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) in PBS for 10 minutes in dark.
The primary antibodies were anti-OCT-4 (1:100; Cell Signaling Technology, Danvers, MA); anti-Nanog homeobox (NANOG) (1:100; Cell Signaling Technology); anti-SOX2 (1:100; Cell Signaling Technology); anti-stage-specific embryonic antigen-4 [SSEA-4] (1:100; Cell Signaling Technology), anti-(tumor rejection antigen [TRA])-1-60 (1:100; Cell Signaling Technology); anti-TRA-1-81 (1:100; Cell Signaling Technology); and antiandrogen receptor (AR) (1:200; Cell Signaling Technology). The secondary antibody was Alexa 488–conjugated goat anti-rabbit IgG (Invitrogen). Fluorescent images were acquired using a Zeiss AXIO Vert.A1 microscope (Zeiss, Germany).
Embryoid body formation and in vitro differentiation
For embryoid body (EB) formation, iPS colonies on Matrigel-coated plates were incubated with ethylenediaminetetraacetic acid (CellAPY, China) for 5 minutes at 37°C until the colonies were completely dissociated. The colonies were washed with Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco), and resuspended in EB medium (Differeasy; CellAPY). The colonies were cultured in suspension for 4 days, and then transferred onto Matrigel-coated plates and cultured for 4 more days.
Teratoma formation
To test the capability of iPSCs to differentiate into structures belonging to the three germ layers, 22RV1-iPSCs (∼1 × 106 22RV1-iPSCs in 100 μL DMEM/F12 medium) were subcutaneously injected into the dorsal flanks of NOD-SCID mice. All of the experiments on mice were approved by the local Ethics Committee of Southern Medical University. About 6 weeks after injection, tumors were noted to form from the 22RV1-iPSC clones. The tumors were dissected at week 8, and tissues were stained with hematoxylin and eosin and examined by light microscopy.
Statistical analysis
Data are expressed as the mean ± standard deviation of three independent experiments. Differences between two groups were examined using a two-tailed Student's t-test. Values of p < 0.05 were considered to be statistically significant. Statistical analyses were performed using SPSS version 20.0 software (SPSS, Inc.).
Results
Generation of iPSCs from prostate cancer cell line 22RV1 cells
The Sendai reprogramming vectors containing the four transcription factors OCT-4, SOX2, KLF4, and C-MYC were transduced into 22RV1 cells. The schedule for human iPSC reprogramming is summarized in Figure 1. After ∼3–4 days, cells infected with the reprogramming vectors began to undergo dramatic morphological changes; from small epithelioid single cells to the formation of cobblestone colonies.

Timeline for 22RV1-derived iPSCs generation. iPSCs, induced pluripotent stem cells.
Approximately 7–12 days after infection, human embryonic stem cell (hESC)-like colonies (tight and flat colonies with well-defined phase-bright borders composed of small cells with a high nucleus-to-cytoplasm ratio) appeared and grew rapidly (Fig. 2A). Approximately 70 identifiable hESC-like colonies from 2 × 105 transduced 22RV1 cells were mechanically picked, and expanded in human iPSC medium on Matrigel. The overall reprogramming efficiency was about 0.03%.
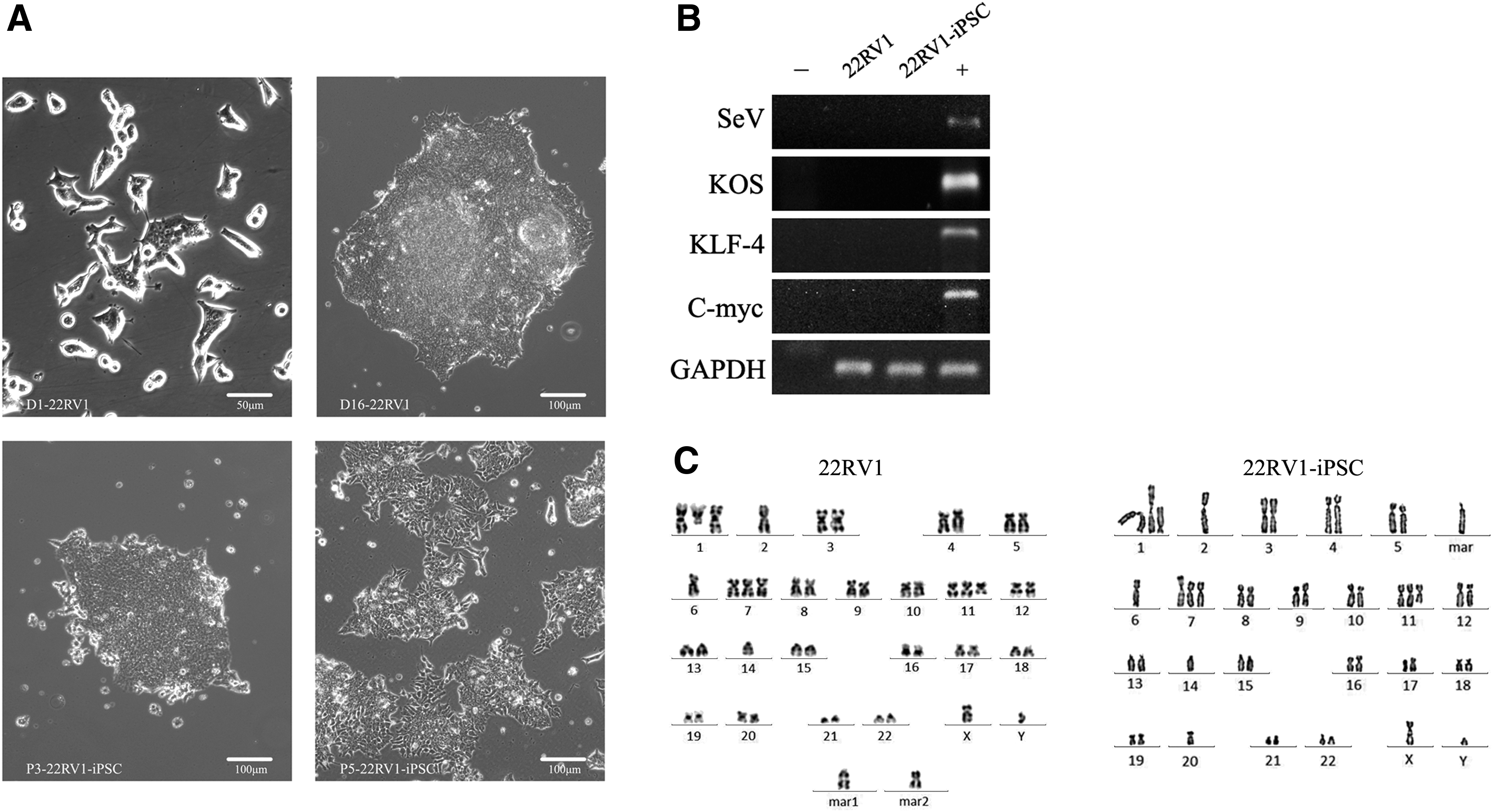
Characterization of a typical nonintegrated iPSC cell line generated from 22RV1.
The Sendai virus system (Sendai virus vector) was used to deliver the reprogramming factors to minimize the potential effects of integrating methods on genomic stability and has been used for iPSC generation from Fabry disease skin fibroblasts (Kawagoe et al., 2013). Using a genomic PCR method that could specifically amplify the transgenes used for reprogramming, it was confirmed that the stably expanded 22RV1-iPSCs at passage six no longer harbored the exogenous reprogramming factors or the Sendai viral backbones (Fig. 2B).
Furthermore, the karyotype of the 22RV1-derived iPSCs [46,XY,del(1)(q24),+der(1)t(1;14)(q10;q10), −2,−6,+7,+del(11)(q14), −14,−20,+mar] were abnormal and not exactly the same as the parental cells [48,XY,del(1)(q24),+der(1)t(1;14)(q10;q10), −2,−6,+7,+del(11)(q14), −14,+2mar]; thus, providing evidence of chromosomal instability (Fig. 2C).
Human 22RV1-iPSCs express hESC-specific genes and markers
Immunofluorescence analysis demonstrated that the human, hESC-specific, surface markers SSEA-4, TRA-1-81, and TRA-1-60, and the transcription factors NANOG, SOX2, and OCT-4 were fully activated (Fig. 3A). Quantitative real-time PCR analysis with primers targeting the endogenous genes OCT-4, NANOG, SOX-2, KLF-4, and C-MYC revealed that the gene expression patterns of the 22RV1-iPSCs were similar to those of the H1 cells (hESC line): NANOG, OCT-4, and SOX2 were expressed at a higher level in 22RV1-iPSC and H1 hESCs, whereas KLF4 and C-MYC were expressed at a lower level (Fig. 3B). In addition, AP activity, typical of an ESC phenotype, was demonstrated in the induced cells (Fig. 3C).

The expression of specific hESC surface markers in the 22RV1-iPSCs.
Human 22RV1-iPSCs are pluripotent and can differentiate into the three germ layers in vitro and in vivo
The EB formation assay to determine the ability of 22RV1-iPSCs to form EBs showed that 22RV1-iPSCs formed typical EBs when cultured in suspension on low-adhesion plates in the absence of basic fibroblast growth factor (FGF) (Fig. 4A) and expressed genes of three germ layer lineages (Fig. 4B).

Pluripotency and differentiation potency of 22RV1-iPSCs.
The ability of iPSCs to differentiate into the three germ layers in vivo was tested by injecting 22RV1-iPSCs into NOD-SCID mice. The results showed that the cells generated teratomas containing tissues of the three germ layers: gland tissue (endoderm), cartilage (mesoderm), and neural rosettes (ectoderm) (Fig. 4C).
22RV1-iPSCs retained expression of AR
Prostate cancer cell line 22RV1 cells express AR; thus, we investigated the expression of AR by 22RV1-iPSCs with 22RV1 cells as control. The immunofluorescence and quantitative RT-PCR (real time-PCR) results indicated that AR was expressed by 22RV1-iPSCs with no differences were observed between 22RV1-iPSCs and 22RV1 cells (Fig. 5A, B), suggesting that prostate cancer-specific cell markers are preserved after transduction to iPSCs.
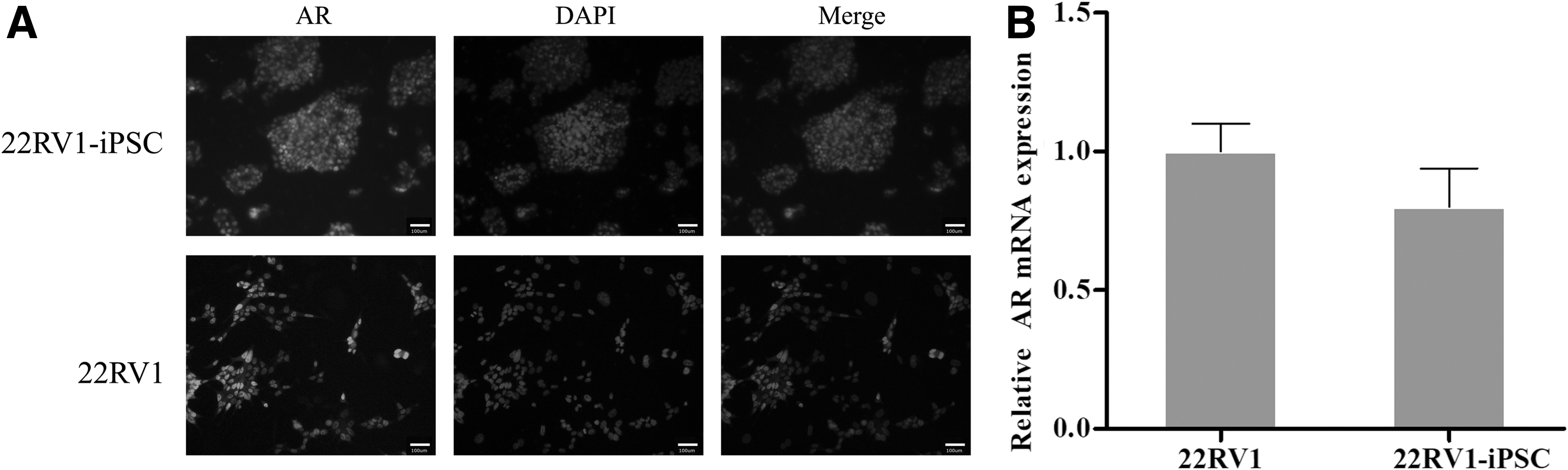
The AR expression in the 22RV1-iPSCs.
Discussion
This study is the first to report the successful reprogramming of human prostate cancer cell line 22RV1 to an ESC-like pluripotent state. The cells were validated as de facto iPSCs by confirming their ability for sustained self-renewal, silencing of exogenous transgenes, expression of ESC-specific genes, and pluripotent differentiation into all three germ lineages, in vitro and in vivo. Furthermore, the expression of functional ARs in 22RV1-iPSCs demonstrated that the 22RV1-iPSCs were transduced from a prostate cancer-specific lineage.
PrCSCs may be a major cause for resistance to therapy, including androgen-deprivation therapy, chemotherapy, and radiotherapy, in advanced prostate cancer. Since the first description of PrCSCs, most studies have focused on the isolation of PrCSCs from human primary prostate cancer cells or prostate cancer cell lines, as well as their biological behavior (Vidal et al., 2014; Zhou et al., 2017). However, the origin of PrCSCs is unknown. Reports have suggested that every adult tissue may have its own stem cells. Thus, one theory of the origin of PrCSCs is that the cells may be derived from immature prostate tissue stem cells or progenitor cells as a result of genetic or epigenetic changes (Romańska-Knight and Abel, 2011).
This theory is supported by the fact that both stem cells and cancer stem cells possess the capacity for self-renewal and unlimited replication, and that stem cells are relatively long-lived in comparison to other cells (Beachy et al., 2004). As a result, the cancer stem cells have a greater opportunity to accumulate multiple additional mutations and this along with an increased rate of proliferation can increase the risk for the development of cancer. In addition, high telomerase activity is detectable in cancer cells and organ- and tissue-specific stem cells and other progenitor cells (Pathak, 2002). Hence, understanding the relationship between cancer stem cells and progenitor cells, and how normal regulation is subverted during the process of carcinogenesis may help develop new therapies for advanced prostate cancer.
Converting cancer cell lines into a pluripotent state may be an effective model for studying the origin of cancer stem cells, and the process by which progenitor cells transform to cancer stem cells.
In 2006, iPSCs technology was developed by Yamanaka et al., who discovered that differentiated adult human cells could be converted to an immature stem cell-like state through induced expression of a combination of transcription factors. This technology allows iPSCs to be generated from various cell types and provides convenient models for studies of normal and diseased biology. Moad et al. (2013) generated human iPSCs from prostate cells and these iPSCs successfully differentiated back into prostate lineages. In addition, Hepburn et al. (2020) found that coculture of human prostate-derived iPSCs with rat UGM cells generated both in vivo xenografts and in vitro prostate organoids that recreated the full breadth in situ prostate epithelial differentiation.
Although a number of studies have reported the successful application of this technology to different types of cancers (Bindhya et al., 2019; Rowe and Daley, 2019), until now neither prostate cancer cell lines nor primary prostate cancer cells have been successfully reprogrammed into iPSCs.
Hepburn et al. (2019) used standard pluripotency/embryonic stem cell media to culture LNCaP cells and found that the morphology of LNCaP cells became similar to that of ESCs and iPSCs with an upregulation of OCT-4, SOX2, and NANOG. However, these cells were not identical to conventional iPSCs. Hence, in our preliminary work, we attempted to transduce several prostate cancer cell lines (LNCaP, PC-3, 22RV1, and DU145) into iPSCs and observed that only the 22RV1 cell line could be successfully converted. While this study has shown that the prostate cancer cell line 22RV1 can be transduced into iPSCs, further research is necessary to determine the mechanisms underlying the transduction.
In conclusion, the results of this study illustrate a method of transducing the prostate cancer cell line 22RV1 into pluripotent 22RV1-iPSCs. This method has the potential to allow investigation into the process of prostate cancer development and the origin and role of PrCSCs in prostate cancer.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was supported by funding from the Natural Science Foundation of Guangdong Province (2017A030310107).