
Abstract
Differentiation of keratinocytes from human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) has become an important tool for wound healing research and for studying skin diseases in instances where patient cells are not available. Several keratinocyte differentiation protocols using hiPSC colony fragments or embryoid bodies have been published with some requiring prolonged time for differentiation or extended use of reagent cocktails. In this study, we present a simplified method to efficiently generate large numbers of uniformly differentiated keratinocytes in less than 4 weeks from singularized hiPSCs with differentiation factors, retinoic acid and bone morphogenetic protein 4 (BMP4). Low seeding density of singularized iPSCs results in keratinocyte cultures with minimum cell death during differentiation and up to 96% homogeneity for keratin 14-positive cells and low percentage of keratinocyte maturation markers, comparable to early passage primary keratinocytes. hiPSC-derived keratinocytes remain in a proliferative state, can be maintained for prolonged periods of time, and can be terminally differentiated under high calcium conditions in the same way as primary human keratinocytes. Moreover, coculturing hiPSC-derived fibroblasts and keratinocytes consistently formed organotypic 3D skin equivalents. Therefore, keratinocytes generated by this method are a viable source of cells for downstream applications.
Introduction
Skin is the largest organ of the human body and consists of epidermis and the underlying dermis. Fibroblasts, the major cell type of the dermal layer, are embedded in a collagenous matrix. Keratinocytes are the major cell type of the epidermis. In the innermost layer of the epidermis, the basal layer, keratinocytes are separated from the dermis by the basal lamina. Basal layer keratinocytes proliferate and undergo a maturation process as they migrate into the outer layers of the epidermis. Characteristic expression markers for proliferating keratinocytes of the inner layer are keratin 14 (KRT14), keratin 5 (KRT5), and tumor protein p63. As keratinocytes mature, they express increasing amounts of differentiation markers, such as keratin 1 (KRT1), involucrin, and filaggrin. Terminally differentiated keratinocytes form the outermost epidermal layer, the stratum corneum, which is important for the barrier function of the skin.
Primary fibroblasts and keratinocytes can be isolated from skin biopsies of patients and are important components for in vitro studies of skin disorders. While fibroblasts from a single biopsy are easily cultured and amplified, primary keratinocytes can be more challenging to amplify and to maintain in sufficient numbers for subsequent cell culture experiments and long-term studies. Patient fibroblasts and keratinocytes are often used for generating organotypic 3D skin equivalents to study rare skin disorders (Itoh et al., 2013), the barrier function of skin (Goleva et al., 2019; Niehues et al., 2018), or wound healing mechanisms (Abaci et al., 2017).
Extensive laboratory investigations with patient-specific skin equivalents require large amounts of patient cells. To repeatedly obtain skin biopsies from the same person may not be possible or may be unethical-especially when investigating rare disorders. For sufficient expansion of primary keratinocyte cultures from a patient for multiple experiments or therapy, complex selection for stem cell-like keratinocytes has been reported (Barrandon and Green, 1987; Hirsch et al., 2017). However, in many cases, this method may not be feasible or economical.
The generation of human induced pluripotent stem cells (hiPSCs) for the purpose of keratinocyte differentiation may be necessary when a continuous source of primary cells is unavailable. This frequently happens in genetic research when functional studies needed to investigate disease-causing roles of putative mutations and patient keratinocytes are not available. hiPSCs have opened up new avenues for medical research to overcome limitations of primary cell cultures. Methods to generate patient-specific hiPSCs were developed to provide a continuous source of stem cells that can then be differentiated into desired cell types (Nakagawa et al., 2008). Genome editing with CRISPR/Cas9 can be used to correct disease mutations or introduce mutations of interest into normal hiPSCs, thus creating isogenic hiPSC lines, which are genetically identical and differ only by the variant of interest (Richardson et al., 2016; Yang et al., 2013).
Establishing hiPSCs from patients is relatively easy and cost-effective but differentiating hiPSCs into specific cell types often remains challenging. A number of protocols have been published for differentiation of human embryonic stem cells (hESCs) and hiPSCs into cell types that are found in skin, such as fibroblasts (Du et al., 2015; Hewitt et al., 2011), keratinocytes (Aberdam et al., 2008; Aberdam, 2004; Guenou et al., 2009; Itoh et al., 2011; Kidwai et al., 2013; Kogut et al., 2014; Metallo et al., 2008; Sebastiano et al., 2014), melanocytes (Hosaka et al., 2019; Ohta et al., 2011), macrophages (Karlsson et al., 2008; Mukherjee et al., 2018), and dendritic cells (Sachamitr et al., 2017; Senju et al., 2011).
Protocols for differentiating hiPSCs into keratinocytes use either stem cell colony fragments or embryoid bodies (EBs) as a basis (Itoh et al., 2011; Sebastiano et al., 2014). Lack of control over cell density in previously published methods can lead to uneven cell overgrowth and resulting cell death, therefore, the time required to obtain homogenously differentiated hiPSC-derived keratinocytes varies between studies.
Challenges with differentiating hiPSCs into keratinocytes include obtaining pure cultures of keratinocytes and large numbers of keratinocytes for downstream experiments. In this study, we modified published protocols (Itoh et al., 2011; Kidwai et al., 2013; Kogut et al., 2014; Sebastiano et al., 2014) to develop a robust method for obtaining highly enriched keratinocyte populations by singularizing hiPSCs and differentiating them under optimized conditions. The resulting hiPSC-derived keratinocytes express the epithelial marker KRT14 in 95% of the cells already after 28 days differentiation and are suitable for long-term expansion to obtain large numbers of keratinocytes.
Materials and Methods
Generation of hiPSC
hiPSCs were derived from peripheral blood of healthy volunteers using integration-free Sendai virus vectors. Generation and use of hiPSCs was approved by our Institutional Review Board (IRB) protocol # 09-199 and the UConn Stem Cell Research Oversight Committee. The hiPSC lines have been tested for pluripotency and genomic integrity (Chen et al., 2013). Three different hiPSC clones were used in this study. Karyotype analysis of G-banded metaphase chromosomes of hiPSCs was performed at the UConn Chromosome Core.
Differentiation of hiPSCs into keratinocytes
hiPSCs were cultured on Matrigel-coated (BD Biosciences) six-well plates with PeproGrow hESC medium (Cat. No. BM-HESC-500; PeproTech) and maintained at 37°C in a humidified atmosphere in the presence of 5% CO2 and 5% O2. For keratinocyte lineage differentiation, we washed hiPSC colonies twice with PBS, singularized with accutase (Cat. No. SCR005; EMD Millipore) for 7 to 10 minutes and stopped the digest with defined keratinocyte serum-free differentiation medium (DKSFM; Gibco). We harvested single cells by centrifugation at 290 g for 5 minutes.
Singularized hiPSCs were resuspended in hESC medium containing ROCK inhibitor (10 μM; Y-27632; Selleck Chemicals) and seeded at a density of 20,000 cells per well on Matrigel-coated six-well plates and incubated at 37°C in a humidified atmosphere in the presence of 5% CO2 and 5% O2. After 24 hours, cells were incubated in DKSFM supplemented with 1 μM all-trans retinoic acid (RA; Cat. No. RA2625; Sigma-Aldrich) and 10 ng/mL of recombinant human bone morphogenetic protein-4 (BMP4; Cat. No. 120-05-ET; PeproTech). DKSFM supplemented with RA and BMP4 was replaced every other day. On day 9, DKSFM containing RA and BMP4 was replaced with CnT-07 medium (CELLnTEC) and cells were fed with CnT-07 medium every other day until day 28.
For passaging of hiPSC-derived keratinocytes (1:3), we washed wells twice with PBS and incubated with 0.25% trypsin/EDTA (Gibco) before centrifugation at 290 g. Cells were maintained in CnT-07 medium containing ROCK inhibitor on collagen-coated dishes (PureCol; Cat. No. 5005; Advanced BioMatrix) and incubated at 37°C in a humidified atmosphere containing 5% CO2 and 21% O2.
Growth curve of hiPSC-derived keratinocytes
We determined the growth curve of hiPSC-derived keratinocytes by counting cell numbers after each passage and performed a cell viability assay for 1–5 days after seeding cells. For growth curve establishment, hiPSC-derived and normal primary keratinocytes were seeded at a density of 5000 cells/cm2 in 10-cm cell culture dishes until a density of 80%–90% was reached. Cells were singularized with accutase and counted with a hemocytometer before each passage. The growth chart over three passages was constructed using the formula: log ((number of cells obtained)/(number of cells seeded))/log (2) (Bisson et al., 2013).
The viability assay was performed by using the CellTiter-Glo 2.0 Luminescent Kit (Promega, Madison). Normal human keratinocytes (NHKs) and hiPSC-derived keratinocytes were seeded at a density of 5000 cells in 100 μL CnT07 medium per well in 96-well plates. Cells were incubated for 1, 2, 3, 4, or 5 days. At each time point, 100 μL of CellTiter-Glo reagent was added to each well. The contents were mixed for 2 minutes on an orbital shaker followed by incubation for 10 minutes in dark at room temperature. Luminescence was measured in a Tecan Safire 2 microplate reader (TECAN, Switzerland).
Differentiation of hiPSCs into fibroblasts
We generated fibroblasts from hiPSCs as previously described (Itoh et al., 2013). Briefly, we generated EBs in AggreWell medium (STEMCELL Technologies) supplemented with 0.3 mM ascorbic acid (Cat. No. A-5960; Sigma), 10 ng/mL transforming growth factor beta 2 (TGF-β2; Cat. No. 100–21; PeproTech), insulin transferrin selenium-A (ITS-A; 1:100; Cat. No. 51300044; Invitrogen) and ROCK inhibitor (10 μM; Y-27632; Selleck Chemicals) on a low binding AggreWell plate (STEMCELL Technologies). We used 1 × 106 hiPSCs to generate 300 EBs.
EBs were transferred to a gelatin-coated six-well plate after 48 hours incubation and cultured in high-glucose DMEM (Cat. No. 11971–025; Gibco) supplemented with 20% fetal bovine serum (Hyclone; GE Healthcare Life Sciences) and 0.3 mM freshly prepared ascorbic acid until confluent. The cells were passaged and cultured until a consistent spindle-shaped cell morphology was observed.
Establishment of 3D skin equivalents with hiPSC-differentiated keratinocytes and fibroblasts
We generated polystyrene scaffold-based 3D skin equivalents following the manufacturer's instructions (Alvetex 3D Cell Culture Technology; AMSBIO). We first seeded 200,000 hiPSC-derived fibroblasts onto an Alvetex scaffold. Fibroblasts were grown in high-glucose DMEM (Invitrogen) for 7 days, followed by seeding 500,000 hiPSC-derived keratinocytes on top of the dermal layer formed by fibroblast embedded into the scaffold. The construct was allowed to mature for 7 days submersed in CnT-07 medium (CellnTech) before airlifting. After airlifting, the medium was changed to CnT-Prime Full-Thickness Skin Airlift medium (CnT-FTAL5; CellnTech) and incubated for another 14 days. Medium was changed every other day during the entire 4-week period. The skin equivalents were harvested for histological analysis.
Flow cytometry
We harvested hiPSC-derived keratinocytes using 0.25% trypsin/EDTA (Gibco) and fixed with 2% paraformaldehyde (PFA; Sigma-Aldrich) for 15 minutes at room temperature. After washing twice with 1 × PBS containing 0.5% BSA and permeabilizing in 0.2% Tween-20 for 15 minutes at 37°C, we blocked with 10% fetal bovine serum for 10 minutes at 4°C and incubated with monoclonal antibodies for fluorescein isothiocyanate-conjugated keratin 10 (KRT10, 1:100; 0.2 μg/μL), phycoerythrin (PE)-conjugated involucrin (IVL, 1:100; 0.2 μg/μL), and allophycocyanin (APC)-conjugated KRT14 (1:100; 0.2 μg/μL) (all from Santa Cruz Biotechnology).
We validated the characteristics of hiPSC-derived fibroblasts by flow cytometry for expression of CD44 (1:100; 0.2 μg/106 cells in 100 μL volume), CD73 (1:100; 0.2 μg/106 cells in 100 μL volume), CD90 (1:100; 0.2 μg/106 cells in 100 μL volume), and CD166 (1:100; 0.2 μg/106 cells in 100 μL volume) surface markers (BioLegend). We performed flow cytometry using a BD LSRII Cell Analyzer (BD Biosciences) and further analyzed the data using FlowJo (Tree Star).
cDNA synthesis and quantitative real-time polymerase chain reaction analysis
We isolated DNaseI-treated RNA using the Direct-zol™ RNA MiniPrep Plus Kits (Zymo Research) and synthesized cDNA with SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. We performed quantitative real-time polymerase chain reaction (qPCR) on a CFX Connect Real-Time PCR Detection System (Bio-Rad) using a SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) with the following human primers:
SRY-Box transcription factor 2 (SOX2), forward 5′-ACACCAATCCCATCCACACT-3′ and reverse 5′-GCAAACTTCCTGCAAAGCTC-3′; Octamer-binding protein 4 (OCT4), forward 5′-GTACTCCTCGGTCCCTTTCC-3′ and reverse 5′-CAAAAACCCTGGCACAAACT-3′; Nanog homeobox (NANOG), forward 5′-GATTTGTGGGCCTGAAGAAA-3′ and reverse 5′-AAGTGGGTTGTTTGCCTTTG-3′; Delta N-type tumor protein p63 (ΔNp63), forward 5′-GGAAAACAATGCCCAGACTC-3′ and reverse 5′-GTGGAATACGTCCAGGTGGC-3′; TA-type tumor protein p63 (TAp63), forward 5′-AAGATGGTGCGACAAACAAG-3′ and reverse 5′-AGAGAGCATCGAAGGTGGAG-3′; Integrin subunit beta 4 (ITGB4), forward 5′-TGGAAGTACTGTGCCTGCTG-3′ and reverse 5′-TGCATGTTGTTGGTGACCTT-3′; Keratin 5 (KRT5), forward 5′-ACCGTTCCTGGGTAACAGAGCCAC-3′ and reverse 5′-GCGGGAGACAGACGGGGTGATG-3′; Keratin 14 (KRT14), forward 5′-GCAGTCATCCAGAGATGTGACC-3′ and reverse 5′-GGGATCTTCCAGTGGGATCT-3′; and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward 5′-CTCTCTGCTCCTCCTGTTCGA-3′ and reverse 5′-TGAGCGATGTGGCTCGGCT-3′.
The amplification protocol consisted of an initial polymerase activation step at 98°C for 30 seconds, followed by 40 amplification cycles at 95°C for 10 seconds and 60°C for 30 seconds. We optimized PCR efficiency and tested primer specificity by melting curve analysis. Expression levels of genes were calculated using the 2−ΔΔCt method and normalized against an endogenous GAPDH control.
Terminal differentiation of hiPSC-derived keratinocytes
We induced terminal differentiation of hiPSC-derived keratinocytes by increasing the extracellular Ca2+ level to 1.27 mM with calcium chloride (Sigma-Aldrich) as previously reported (Pillai et al., 1990; Xie et al., 2005). Briefly, we cultured keratinocytes at a density of 0.5 × 106 cells/4 mL CnT-07 medium (containing 0.07 mM calcium) in collagen-coated 60-mm culture dishes without the addition of ROCK inhibitor. Cells were cultured in 1.27 mM Ca2+ when they reached ∼80%–90% confluency for 2–3 days. Expressions of KRT1, loricrin, and filaggrin, markers of terminally differentiated keratinocytes, were evaluated by immunoblots.
Immunoblot analysis
We prepared whole cell lysates from differentiated keratinocytes and fibroblasts using RIPA buffer. Equal amounts of protein determined by BCA assay (Pierce) were loaded for each lane. For keratinocyte analysis, the membranes were incubated overnight with antibodies against KRT1 (1:1000), loricrin (1:1000), filaggrin (1:1000), and GAPDH (1:1000). Immunoblots with fibroblast lysates were incubated with fibroblast-specific protein (FSP1) (S100A4; 1:1000) and vimentin (1:1000). All antibodies were purchased from Santa Cruz Biotechnology.
Antibodies were diluted in Tris-buffered saline containing 0.1% Tween-20 and 5% skim milk. After incubation with primary antibodies, the membranes were washed in Tris-buffered saline/Tween buffer and then incubated with peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (Azure Biosystems) secondary antibodies (1:10,000 in Tris-buffered saline/Tween buffer containing 5% skim milk) for 1 hour. Bound antibodies were detected using a Radiance chemiluminescence system (Azure Biosystems).
Immunofluorescence analysis
Cells seeded on cover slips were fixed in 4% PFA. Immunofluorescence labeling was performed as previously described (Wojcik et al., 2001). Primary antibodies against NANOG (ab109250) and SOX2 (ab137385) were purchased from Abcam, and OCT3/4 (sc-5279), KRT14 (sc-532553), and KRT18 (sc-323239) from Santa Cruz Biotechnology. The secondary antibody conjugate were Alexa-conjugated fluorochrome 488 Donkey Anti-Rabbit (A21206) or Anti-mouse (A21202) from Molecular Probes.
Immunohistochemistry
Three-dimensional skin constructs were placed in 30% sucrose/PBS for 3–4 hours at room temperature before embedding in OCT frozen tissue section medium (Fisher Scientific). Samples were sectioned at a thickness of 20 μm by cryotome (Leica). Histology sections were fixed in 2% PFA for 10 minutes at room temperature before H&E staining. To analyze keratinocyte distribution, sections were stained with a pan-cytokeratin (AE1/AE3) monoclonal antibody (1:50 in 0.1% Tween-20; Cat. No. 914202; BioLegend) after fixing in 2% PFA, permeabilization with 0.1% Triton X-100 in PBS, and blocking in 1% BSA. Colorimetric detection was done by HRP-conjugated secondary antibody (Vector Labs), DAB chromogen solution (Vector Labs), and Hematoxylin counterstaining.
Results
hiPSC-derived keratinocyte differentiation and maintenance
We established a methodology for keratinocyte differentiation from hiPSCs with improved consistency and reproducibility by modifying published protocols. A schematic of our protocol to differentiate hiPSCs into keratinocytes is shown in Figure 1A. We used three clones of hiPSCs derived from peripheral blood of a white female in the third decade of life, which had been carefully characterized and successfully used for differentiation into myeloid cells (Chen et al., 2013, 2017).
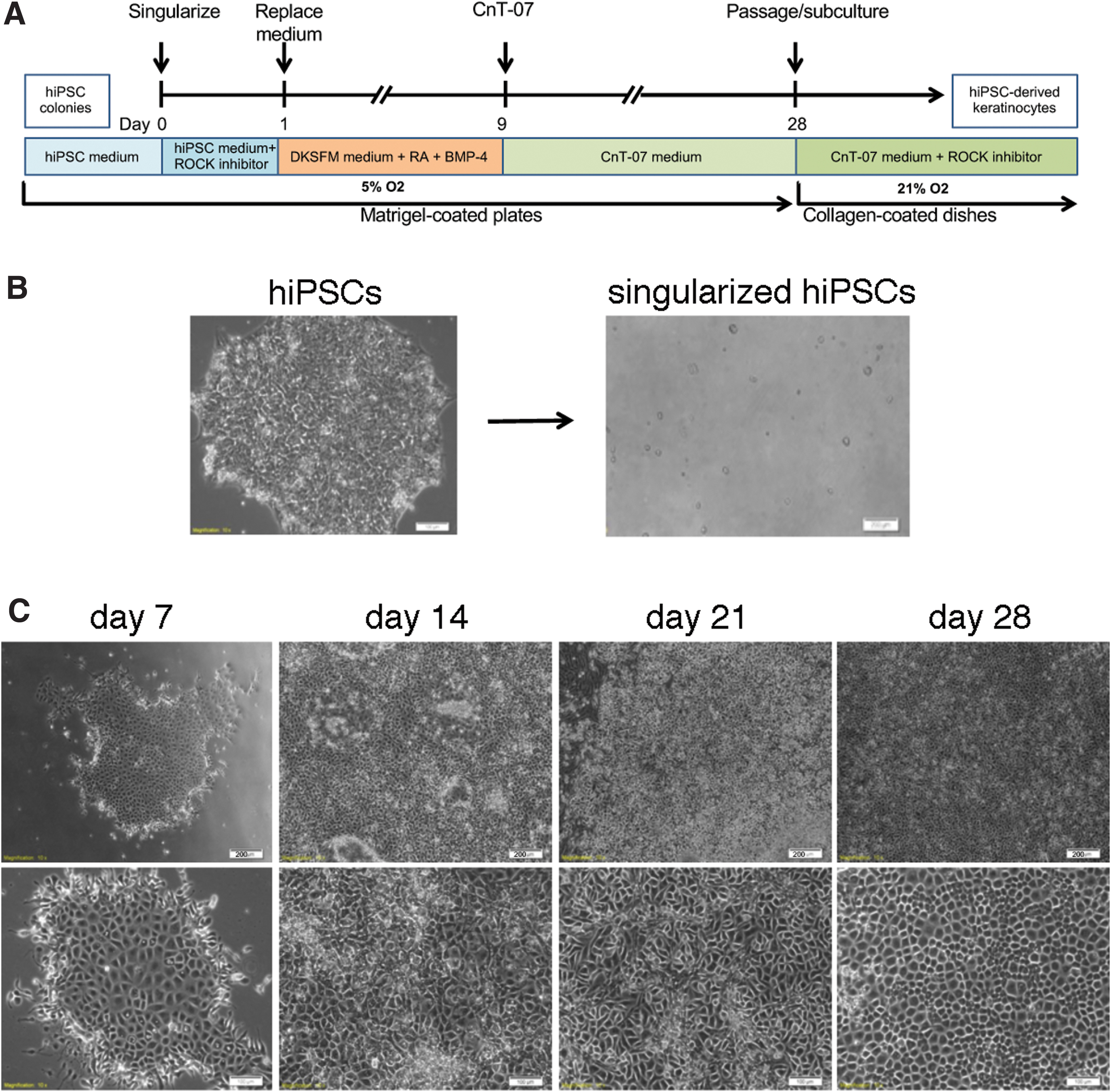
Differentiation of hiPSCs into keratinocytes under defined culture conditions.
Different from the previously published methods, which used EBs or clusters of hiPSCs, we singularized hiPSC colonies and seeded cells in the presence of ROCK inhibitor onto Matrigel-coated six-well plates (Fig. 1B). Major differences between our approach and previous differentiation strategies are summarized in Table 1. We tested seeding densities of 10,000, 20,000, and 30,000 singularized hiPSCs per well of a six-well plate. A seeding density of 20,000 cells per well was optimal for the hiPSC clones that we tested.
Comparison of Keratinocyte Differentiation Protocols
BMP4, bone morphogenetic protein-4; DKSFM, defined keratinocyte serum-free medium; EGF, epidermal growth factor; FAD, F12, adenine and DMEM; FBS, fetal bovine serum; hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell; RA, retinoic acid.
After 24 hours, we switched from hESC medium to DKSFM supplemented with RA and BMP4 to block neural fate and induce ectodermal fate, respectively. On day 9, we switched to CnT-07, a keratinocyte growth medium, and replaced medium every other day until day 28. Differentiating cells had acquired cobblestone-shaped cell morphology by day 7 (Fig. 1C). We observed that differentiation at low oxygen conditions (5% O2) resulted in more consistent cell growth and differentiation.
We passaged the cells (1:3) onto collagen-coated plates when they reached confluence, approximately at day 28. The hiPSC-derived keratinocytes can be further enriched and expanded for longer time when subcultured in keratinocyte medium (CnT-07) supplemented with ROCK inhibitor to increase and maintain their proliferation potential (Chapman et al., 2014; Strudwick et al., 2015). We have passaged hiPSC-differentiated keratinocytes successfully at least nine times.
We found remarkably less cell death in keratinocyte differentiation cultures with single cell seeding than in cultures using cut hiPSC colony fragments or EBs, which led to localized high cell density around colony fragments. Supplementary Figure S1 shows examples of such failed attempts using colony fragments. After initial differentiation into cobblestone-shaped cells, we experienced subsequent crowding around the seeded hiPSC fragments. In these cultures, many cells differentiated into fibroblastoid or neuron-like cell types around cell clusters and by week 2 or 3 of culture we began to see extensive cell death with few cobblestone-shaped cells surviving.
Characterization of hiPSC-derived keratinocytes
Next, we characterized hiPSC-derived keratinocytes at 7, 14, 21, and 28 days of differentiation using keratinocyte markers KRT14, IVL, and KRT10 by flow cytometry. hiPSCs were differentiated into a homogeneous population of KRT14-positive cells, with 67% ± 13.43% positive cells at day 7 and 73% ± 2.73%, 88% ± 2.24%, and 95% ± 2.63% positive cells at days 14, 21, and 28, respectively (Fig. 2A, B).
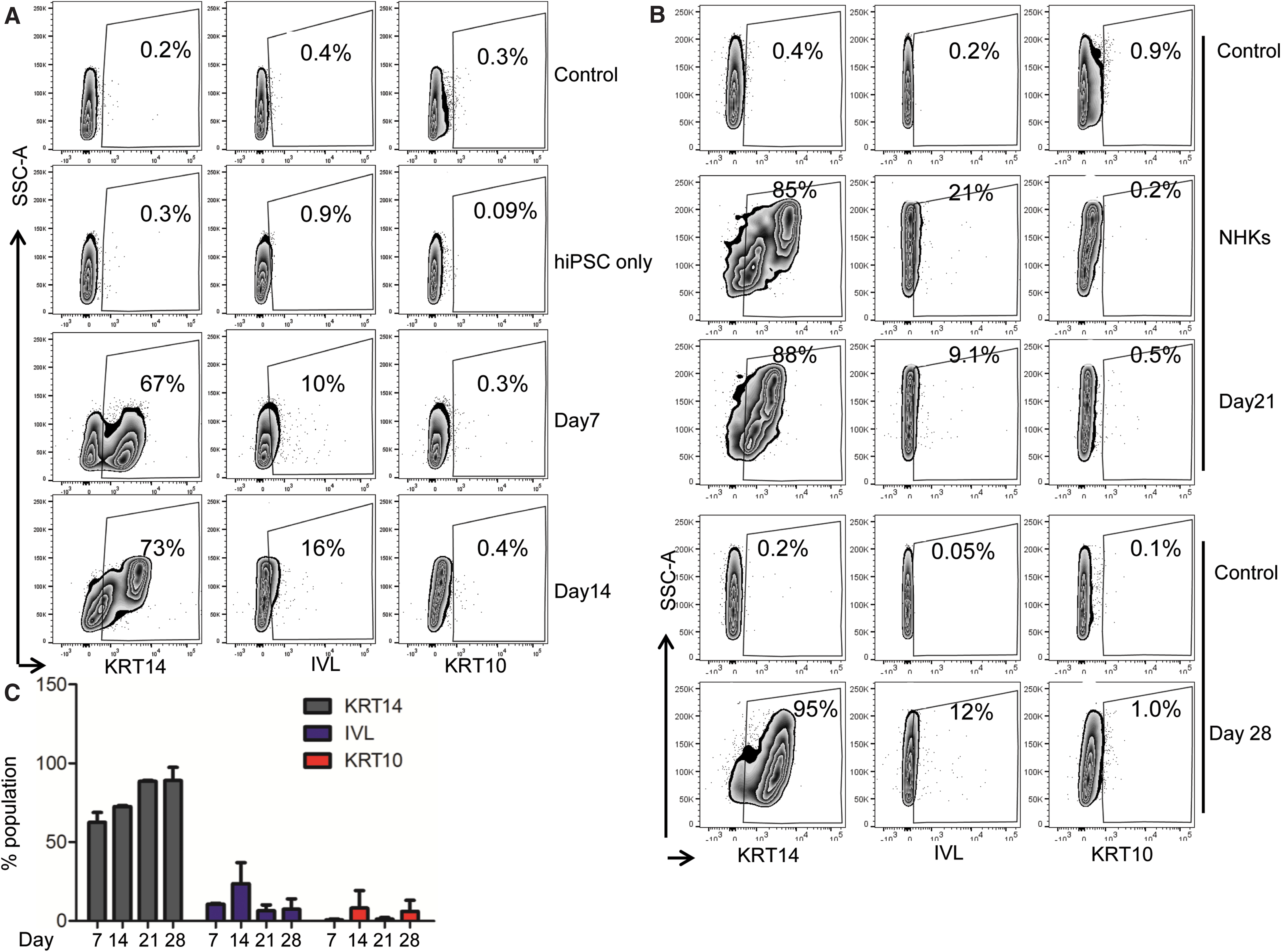
hiPSC-derived cells highly express early basal keratinocyte-specific marker KRT14.
Maturation and terminal differentiation markers, such as IVL and KRT10 levels remained low compared with KRT14 levels, suggesting that differentiating keratinocytes remain in a proliferative mode during the first 28 days of differentiation (Fig. 2A–C). After several passages, hiPSC-derived keratinocytes displayed a homogenously high expression of KRT14 and low levels of IVL and KRT10, similar to cells on day 28, suggesting that hiPSC-derived keratinocytes can maintain their proliferating state in vitro.
Fully differentiated (day 30) hiPSC-derived keratinocytes have a relative growth rate and cell proliferation rate comparable to normal human adult primary keratinocytes as determined by a proliferation assay over 3 passages and a cell viability assay over 5 days (Supplementary Fig. S2).
We examined differentiating hiPSC-derived keratinocytes with characteristic stem cell and keratinocyte markers by qPCR. The differentiation commitment to an ectodermal fate was observed in early stages of differentiation by KRT18, and was drastically reduced in the final differentiation processes by day 28 (Fig. 3A). By day 7, hiPSC-derived keratinocytes expressed not only KRT5 and KRT14 but also other characteristic epidermal and keratinocyte markers, such as ΔNp63, TAp63, and ITGB4, indicating that these cells closely resemble NHKs (Fig. 3A).
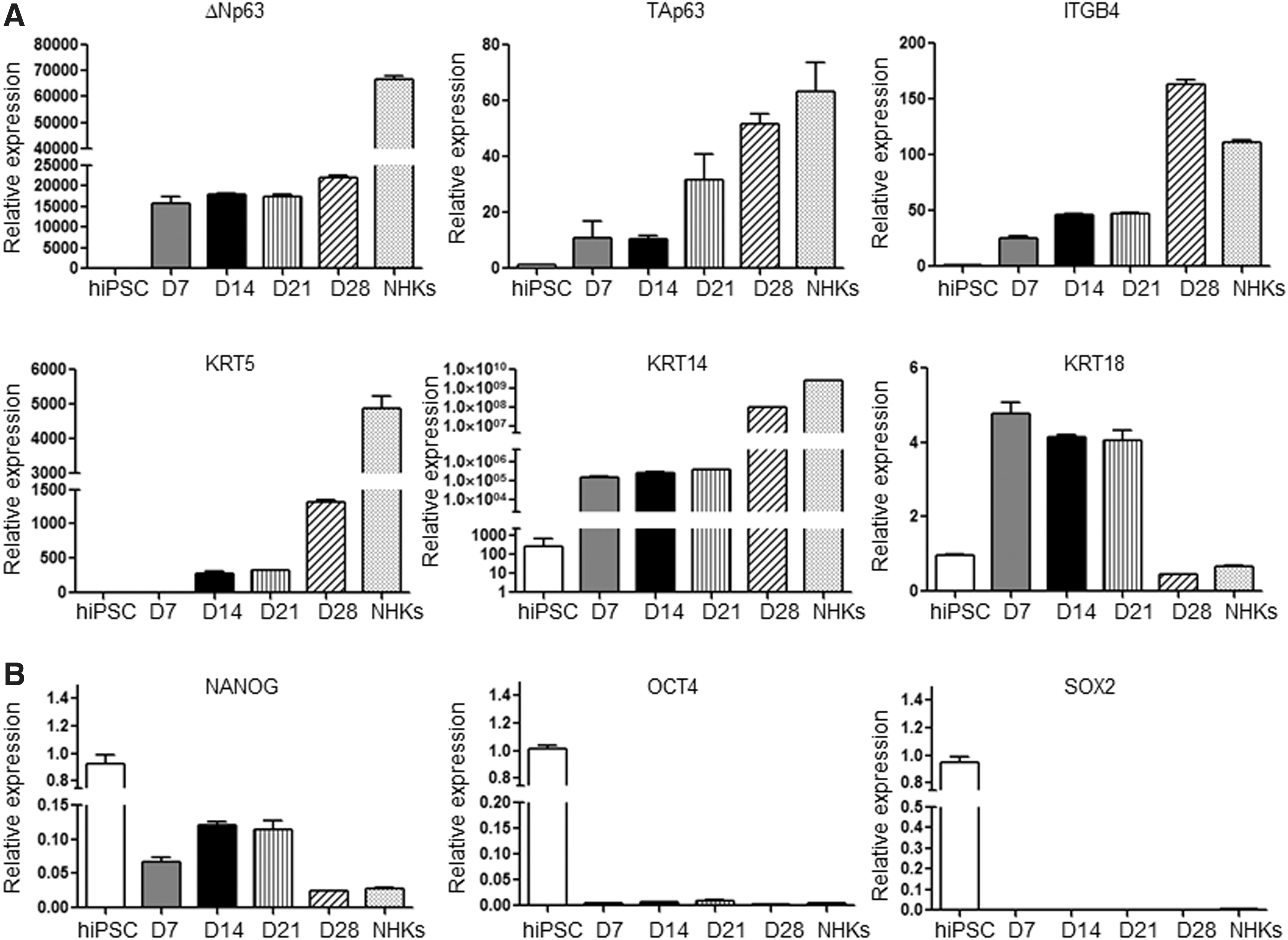
Characteristics of hiPSCs and hiPSC-derived keratinocytes.
On the other hand, the expression levels of stem cell markers, NANOG, OCT4, and SOX2, were greatly reduced or became undetectable in hiPSC-derived keratinocytes (Fig. 3B). This finding was further confirmed by immunofluorescence staining. KRT14 was homogenously and strongly expressed in hiPSC-derived keratinocytes, comparable to NHKs, whereas KRT18 was weakly expressed (Fig. 4A). Furthermore, NANOG, OCT4, and SOX2 were only expressed in hiPSCs but undetectable in hiPSC-derived keratinocytes (Fig. 4A, B).

Immunofluorescence staining of iPSC-derived keratinocytes expressing keratinocyte but not stem cell markers.
Terminal differentiation of hiPSC-derived keratinocytes
Keratinocytes can differentiate and stratify in response to increased extracellular calcium concentration (Pillai et al., 1990; Xie et al., 2005). Therefore, we examined whether hiPSC-derived keratinocytes can differentiate into mature keratinocytes when stimulated by a high calcium concentration (1.27 mM calcium chloride supplementation) in vitro. Regular culture medium contained 0.07 mM calcium.
Our data showed that hiPSC-derived keratinocytes begin to flatten and stratify after 2 days in high calcium conditions (Fig. 5A, B) and cells express higher levels of KRT1, loricrin, and filaggrin as shown by immunoblots (Fig. 5C). These data, taken together, showed that hiPSC-derived keratinocytes can be terminally differentiated under increased extracellular calcium conditions and their response to high calcium stimulation resembles that of NHKs.

Differentiation of hiPSC-derived keratinocytes in response to high extracellular calcium. Brightfield images of hiPSC-derived keratinocytes under the treatment of low (0.07 mM) and high (1.27 mM) extracellular calcium concentrations
Characterization of hiPSC-derived fibroblasts
We cultured hiPSC-derived fibroblasts for 60 days before analyzing for mesenchymal markers CD44, CD73, CD90, and CD166 according to a previously published protocol (Itoh et al., 2013). FACS analysis of these spindle-shaped cells showed that expression of mesenchymal markers in hiPSC-derived fibroblasts is comparable to normal primary human skin fibroblasts (Supplementary Fig. S3A, B). hiPSC-derived fibroblasts also express the intermediate filament protein vimentin and the S100 calcium-binding protein A4 (S100A4), also known as FSP1, suggesting that these cells are fibroblasts (Supplementary Fig. S3C).
Establishment of 3D skin equivalents using hiPSC-derived skin cells
Three-dimensional skin equivalents have been established to study wound healing and skin disorders in vitro (Itoh et al., 2013; Prunieras et al., 1983). In this study, we studied whether our hiPSC-derived keratinocytes and hiPSC-derived fibroblasts can also be differentiated to organize in skin equivalents using a commercially available Alvetex scaffold. Alvetex, an inert porous polystyrene material, offers some advantages for easy culturing and data analysis, including histological sectioning and staining.
Our data showed that hiPSC-derived keratinocytes formed a layered epidermis with a cornified layer on top of the dermal layer derived from hiPSC-derived fibroblasts shown by H&E staining (Fig. 6A). Pan-cytokeratin-positive cells were restricted to the epidermal layer, suggesting the presence of a pure population of keratinocytes (Fig. 6B). Therefore, hiPSC-derived keratinocytes provide a rather homogenous source for the generation of full-thickness artificial skin equivalents.
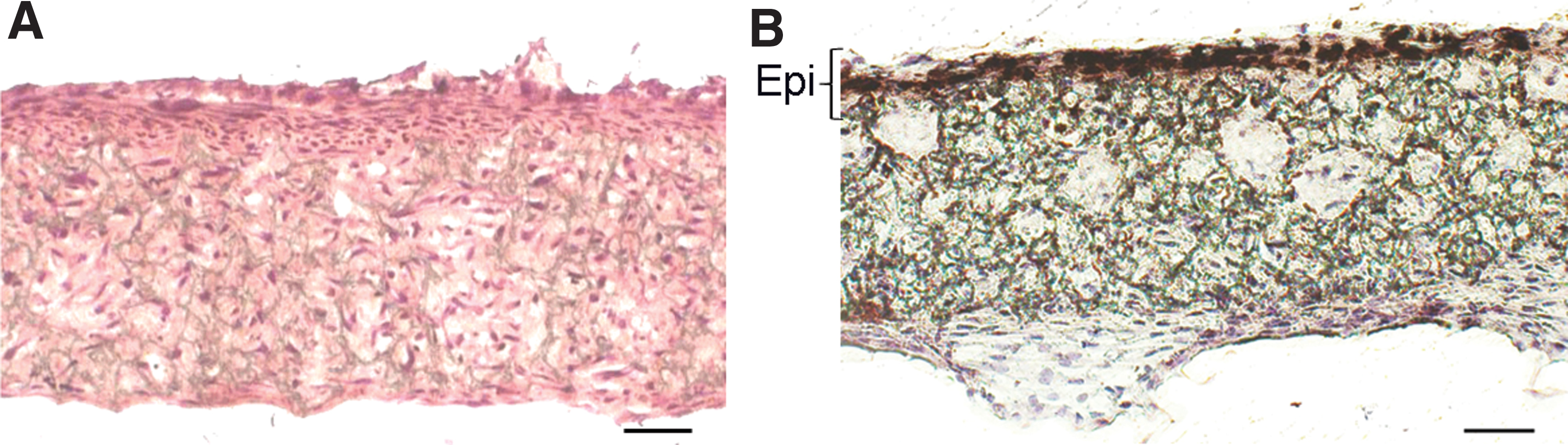
Generation of 3D organotypic skin equivalent using iPSC-derived keratinocytes and fibroblasts.
Discussion
In this study, we present a streamlined approach for effective and reproducible differentiation and propagation of keratinocytes from hiPSCs with important details described. We directly differentiated hiPSCs in a feeder-free and serum-free defined condition within a relatively short timeline. For better control over cell density and more consistent differentiation results, we used singularized hiPSCs instead of EBs or hiPSC fragments.
The initial seeding density is important as cell-cell interaction may affect the quality of keratinocytes during differentiation (Movahednia et al., 2015). Plates where we seeded singularized cells displayed significantly less cell death than plates using cut hiPSC colony fragments or EBs, which developed high cell density around colony fragments. Those cell clusters had a tendency to become crowded and differentiated into elongated cells or neuron-like cells when cultured in DKSFM and CnT-07 (Supplementary Fig. S1). After 2–3 weeks, we found increasing cell death in the culture with only small clusters or individual cobblestone-shaped cells surviving.
In contrast, singularized cells uniformly differentiated into cobblestone-shaped cells, proliferated, and evenly populated the culture plate (Fig. 1).
While keratinocyte selection in other protocols takes up to 3 months (Sebastiano et al., 2014), our approach allowed for obtaining full 10-cm cell culture dishes of ∼95% KRT14-positive keratinocyte-like cells with uniform cobblestone cell shape in less than 1 month. Even at day 21, 88% ± 2.24% of cells are already KRT14 positive. Kidwai et al. (2013) differentiated hESCs through EBs into keratinocytes by stimulation with RA for 10 to 25 days, where 78% of keratinocytes became KRT14 positive after 30 days and 96% positive after 10 passages.
Itoh et al. (2011) used a combination of RA and BMP4 to induce ectodermal fate and inhibit neural differentiation on hiPSC clumps for the initial 4 to 8 days. Those hiPSC-derived keratinocyte cultures reached 45% KRT14-positive cells after 30 days of differentiation and 71% after the first passage (Itoh et al., 2011). Another study reported a 96% KRT14 positivity rate at day 60 of keratinocyte differentiation (Jackow et al., 2019). Characteristics of keratinocyte differentiation methods are summarized in Table 1.
Several protocols suggest differentiating hESCs or hiPSCs into keratinocytes on a fibroblast feeder layer with various combinations of serum, small molecules, and growth factors (Anderson et al., 2018; Chapman et al., 2010, 2014). One study used more elaborate steps, including hiPSCs cultured on feeder cells, EB formation, induction with RA, and subsequent differentiation and keratinocyte selection with RA and BMP4 supplementation for 60 additional days to obtain keratinocytes that could be grafted onto mice (Sebastiano et al., 2014). In another approach, TGF-β receptor inhibitor SB431542, BMP4, and ascorbic acid have been used for the entire 30 days of hiPSC-derived keratinocyte differentiation (Shalom-Feuerstein et al., 2013).
Many protocols for differentiating hESCs and hiPSCs use FAD (F12, adenine, DMEM) medium (Allen-Hoffmann and Rheinwald, 1984) for initiation of ectodermal differentiation together with RA to block neural fate and BMP4 to induce ectodermal fate. While this initiation medium (based on DMEM and F12 media) contains ∼1 mM calcium, other groups, including our group, used keratinocyte growth medium (DKSFM) for initiation, which has a calcium concentration of 0.1 mM (Itoh et al., 2011, 2013; Kogut et al., 2014). It appears that the calcium concentrations of both media are tolerated by hiPSCs and hESCs and that calcium concentration is not a determining factor for inducing ectodermal differentiation. However, low calcium concentration supports keratinocyte proliferation. Therefore, virtually all protocols use media, such as DKSFM and CnT-07, for subsequent keratinocyte differentiation.
Alternatively, hiPSC-derived keratinocytes can be differentiated from skin-derived precursor cells (SKPs). SKPs are differentiated from hiPSCs through a multipotent neural crest-like stage by differentiation initiation with noggin to block BMP signaling and an inhibitor for TGF-β signaling (SB431542), followed by an inhibitor of WNT signaling (CHIR99021). These cells are multipotent and can be differentiated into fibroblasts, keratinocytes, and hair follicles, similar to SKPs isolated from skin (Sugiyama-Nakagiri et al., 2016).
Other studies suggest that keratinocyte-like cells can be obtained by direct conversion of human fibroblasts by transfection with p63, a regulator of epithelial development and KLF4, which regulates epidermal differentiation of these keratinocyte-like cells. However, those cells failed to stratify on dermis, suggesting that they were only partially differentiated (Chen et al., 2014).
Kurita et al. (2018) reprogrammed human fibroblasts, adipose-derived stromal cells and wound-resident mesenchymal cells by overexpressing four factors (NP63A, GRHL2, TFAP2A, and MYC) to generate skin epithelial tissue. Keratinocytes that formed under these experimental conditions expressed KRT13 instead of KRT14. KRT13 is characteristic for mucosal keratinocytes. These examples show that direct conversion methods are promising but still need improvement.
To maintain the proliferative capacity of hiPSC-derived keratinocytes, we cultured them with Y-27632 (ROCK inhibitor) after the 28-day differentiation period. Y-27632 inhibits the serine/threonine Rho-associated protein kinases 1 and 2 (ROCK1, ROCK2), which are downstream targets of the small GTPase Rho. While ROCK1 and ROCK2 have opposite effects on keratinocyte differentiation, it is mainly the inhibition of ROCK2 that prevents terminal differentiation of keratinocytes. ROCK2 promotes terminal differentiation of keratinocytes, whereas ROCK1 acts inhibitory on terminal differentiation of keratinocytes (Lock and Hotchin, 2009).
Y-27632 can facilitate cell adhesion and survival when passaging iPSC lines (Claassen et al., 2009). It stabilizes the telomere length of proliferating keratinocytes (Chapman et al., 2010). Y-27632 has also been used to prolong proliferative properties of primary keratinocytes from skin biopsies (Anderson et al., 2018). Keratinocytes cultured with Y-27632 retain their capacity for terminal differentiation and can be used for subsequent differentiation experiments such as organotypic skin equivalents (Strudwick et al., 2015). In the presence of ROCK inhibitor, we were able to expand our hiPSC-derived keratinocyte cultures for at least nine passages.
Chapman et al. (2010) report that primary human keratinocyte cultures can be passaged for up to 150 times in the presence of ROCK inhibitor. It appears that the presence of ROCK inhibitor increases proliferative properties of keratinocytes by downregulating differentiation through increased AKT and ERK signaling and reduced TGF-β signaling (Ligaba et al., 2015). Primary cultures of human keratinocytes without ROCK inhibitor typically decline after four to five passages.
In summary, while expansion of patient keratinocytes for clinical or research purposes is possible if cells are available, there are many situations where patient cells are not available. hiPSCs may still be the best approach in genetic research for gene editing and differentiation of mutant hiPSCs into keratinocytes, fibroblasts, and other skin-resident cells to ensure continuous supplies of cells. The detailed protocol we present in this study for hiPSC-derived keratinocyte differentiation is a modification and optimization of several previously published protocols for efficient and reproducible keratinocyte differentiation.
Footnotes
Acknowledgments
The authors are grateful to Dr. David Owens and the Skin Stem Cell and Manipulation Core at Columbia University for helpful discussions and assistance.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was supported by the National Institutes of Health (NIH) through the National Center for Research Resources grant M01RR006192 to the Clinical Research Center at UConn Health, and National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR45286 to E.R.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.