
Abstract
Although the molecular pathogenesis of hepatocellular carcinoma (HCC) is uncertain, it is known that the epithelial–mesenchymal transition (EMT) mechanism and epigenetic changes have an important role. This study was focused on evaluating the relationship of 3-Deazaneplanocin A (DZNep) with the EMT mechanism, which is a histone methyltransferase inhibitor on HCC and is also known as an enhancer of zeste homolog 2 (EZH2) inhibitor. Cell viability of HepG2 cells (HCC cell line) assessed for DZNep over 72 hours with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Additionally, colony-forming assay, apoptosis assay, RNA isolation, cDNA synthesis, and real-time PCR (RT-PCR) were performed to see the effect of DZNep on HepG2 cells. DZNep reduced cell proliferation for 72 hours, also significantly reduced colony formation in addition it increased the total apoptosis. DZNep on EZH2, E-cadherin, N-cadherin, and Vimentin (Vim) gene expressions was given different results by either decreasing or increasing the expressions. In this study, we observed a positive effect of DZNep on apoptosis and TIMP3 expression level and decreased colony formation. However, it gave complicated results with the level of gene expression E-cadherin and TIMP2, increase the level of Vim and MMP2 expression. Therefore, we think that further studies are necessary to clarify the role of DZNep.
Introduction
Hepatocellular carcinoma (HCC) is a poor prognosis disease as the primary type of liver cancer and ranks third among the causes of death due to cancer (Li et al., 2019).
The molecular pathogenesis of HCC is uncertain, however, epithelial–mesenchymal transition (EMT) mechanism and epigenetic changes are also known to play an important role (Fan et al., 2019). It is critical to determine how epigenetic modifications such as methyltransferase and demethylase enzymes are included in specific histone targets in HCC (Han et al., 2018).
The C-terminal SET domain of enhancer of zeste homolog 2 (EZH2) exhibits the methyltransferase activity. EZH2 suppresses gene expression by methylating lysine 27 of histone 3 (H3K27) (Tan et al., 2014). Recent studies have shown that EZH2 has physical and functional linkages that suggest a potential interaction between these different classes of epigenetic modification enzymes in the control of gene expression between DNA methyltransferases (DNMTs) and histone deacetylases (HDACs) (Chen et al., 2016; Kouzarides, 2002; Yoo and Jones, 2006).
In recent years, EZH2 has been reported to be associated with many malignant tumors and increased in cancers, such as breast, lymphoma, myeloma, colorectal, and endometrial cancer (Chang and Hung, 2012). Therefore, inhibition of EZH2 has been targeted by epigenetic mechanisms by considering that it plays an oncogenic role in cancer. Inhibition of EZH2 through both methylation inhibitors and histone deacetylation inhibitors has been reported to reduce proliferation and colony formation of cancer cells and induce apoptosis (Huang and Ling, 2017; Shen et al., 2019). On the contrary, recent studies are reporting that EZH2-mediated inhibition of H3K27me3 increases the invasion of breast, ovarian, and pancreatic cancers (Wei et al., 2008). Therefore, the role of EZH2 is uncertain in cancer.
EMT is an important process in which epithelial cells acquire mesenchymal properties, reduce intracellular adhesion, and increase invasive features and mobility (Lamouille et al., 2014). During the EMT process, epithelial cancer cells gain invasive features by acquiring a mesenchymal phenotype (Larue and Bellacosa, 2005). Studies on the role of EMT in the progression of many types of cancers. such as HCC are gradually increasing (Mima et al., 2013).
E-cadherin (E-cad) is the most important marker of EMT in cancer. E-cad is a cell adhesion molecule that directs metastasis, and a decreased expression level leads to increased invasion and metastasis in cancer (Kalluri and Weinberg, 2009; Liu et al., 2014). E-cad expression level was found to be low in many cancer cells. Vimentin (Vim) is one of the important mesenchymal markers in EMT and decreased Vim expression has been reported to reduce the migration and invasion capacity of cancer cells (Liu et al., 2015).
There are conflicting publications about the effect of EZH2 inhibition on the EMT pathway. Inhibition of EZH2 has been reported to inhibit EMT in head and neck squamous, breast, and esophageal cancers (Chang et al., 2016). Also, it has been reported that EZH2 inhibition activates the EMT pathway and cancer cells gain tumorigenic and metastatic properties (Cardenas et al., 2016). Therefore, the role of EZH2 on the EMT pathway is still controversial (Lachat et al., 2019).
Disruption of the normal physiological structure of extracellular matrix (ECM) components causes various liver diseases. Matrix metalloproteinases (MMPs) are the main enzymes involved in ECM destruction (Scheau et al., 2019).
MMPs change during the EMT in many malignancies, such as HCC, and lead to the induction of the EMT process, causing an increase in invasion and metastasis of cancer cells (Giannelli et al., 2002).
MMP-2 is not normally found in liver cells but is expressed in HCC cells, especially in the fibrolamellar variant (Zhang and Chen, 2017). Inhibitors of MMPs (TIMPs, TIMP1-4) play a central role in regulating MMP activity and the deregulation of TIMPs between MMPs promote many human diseases, including cancers (Perigny et al., 2008).
Our study is the first study to evaluate the relationship of 3-Deazaneplanocin A (DZNep) with the EMT mechanism, which is a histone methyltransferase inhibitor on HCC and is also known as an EZH2 inhibitor. In the present study, DZNep was used for inhibition of EZH2 in the HCC cell line (HepG2), then it was aimed to investigate the effect of inhibition of EZH2 on E-cad, Vim, MMP2, TIMP1-3, and genes (EMT and ECM genes) in HepG2 cells. At the same time, the effect of EZH2 inhibition on cell proliferation, colony formation, and apoptosis were also investigated.
Materials and Methods
Ethical review and approval were waived for this study since the study did not involve humans or animals.
Chemical
HepG2 was obtained from the American Type Culture Collection (ATCC). DZNep was obtained from Cayman Chemical. Dulbecco's modified Eagle's medium (DMEM) (low-glutamine), penicillin/streptomycin, and glutamine were obtained from Biological Industries (Kibbutz Beit Haemek 25115 ISRAEL). Dimethyl sulfoxide (DMSO) was obtained from Merck (Merck KGaA, Darmstadt, Germany). Count & Viability and Annexin V & Dead Cell Kits were obtained from Merck Millipore (Billerica, MA, cat. no. MCH 100102, cat no. 100105). TRIzol reagent was obtained from Thermo Fisher Scientific (cat. no. 15596026).
Cell lines and culture conditions
We used HepG2 cells and were cultured in DMEM Low, which was supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin/streptomycin. The stock concentration of DZNep was determined as 5 μM and DMSO (BioChemical) was used as the negative control. HepG2 cells were cultured in 6-well plates in triplicate and 5 μM DZNep applied for 72 hours. We formed two groups of nontreated (control group 1) and treated groups (treated with DZNep group 2). Ethics Committee approval is not needed.
MTT assay
HepG2 cells were cultured (8 × 103 to 10 × 103 per cell) in a 96-well plate in DMEM Low complete. After 24 hours of incubation, cells were treated with different concentrations (0.1, 0.5, 1, 2, 2.5, 5, 7.5, and 10 μM) for 72 hours DZNep. Then 20 μL of MTT solution was added in each well and incubated for 2–4 hours until formazan crystals formed. The crystals were dissolved with DMSO. It is incubated in the dark for15 minutes. Cells were measured with enzyme-linked immunosorbent assay (ELISA) reader.
Cell viability assay
HepG2 cells (3 × 105 per well) were cultured in DMEM Low Complete, and after 24 hours of incubation, cells were treated with time-dependent (24, 48, 72, 96, and 120 hours) DZNep. Then cells were washed with phosphate-buffered saline (PBS) twice and harvested by trypsin. HepG2 cells were centrifuged at 300 g for 5 minutes. The samples were stained with 450 mL Count & Viability Assay Kit at room temperature for 5 minutes in darkness. Then, cell viability was analyzed using flow cytometry. The IC50 value was calculated by GraphPad Prism Software. The IC50 value was determined as to be 5 μM according to this formula;
IC50 = 100 − [(control − treated group)/control −100] (control is cell number in the nontreated group, the treated group is the number of cells in the DZNep-treated group at different concentrations).
Colony-forming assay
HepG2 cells were cultured (100 per cell) in a 6-well plate and DZNep-treated group was treated with 5 μM DZNep. After 24 hours, the medium was replaced until colony formation was observed (9 days) and cells were washed with dPBS. Cells (each plate) were stained with 10% Crystal Violet and after colony formation was observed, cells were photographed and counted.
Apoptosis assay
HepG2 cells were cultured, 3 × 105 cells per well in a 6-well plate. The DZNep-treated group was treated with 5 μM DZNep for 72 hours. Cells were harvested with trypsin, subjected to centrifugation at 350 g for 5 minutes, then washed once using PBS. The cells were incubated for 20 minutes by adding PBS mixture with the appropriate kit and flow cytometry was used to measure. All experiments were done in triplicate.
RNA isolation, cDNA synthesis, and real-time PCR (RT-PCR)
HepG2 cells were cultured in a culture flask, then cells were treated with DZNep for 72 hours. After 72 hours of incubation, medium was aspirated. The cell was washed with PBS, then cells were harvested using TRIzol reagent and cell scraper. RNA isolation was analyzed by the chloroform/phenol method. RNAs were measured by Nanodrop, then the cDNA synthesis was performed using the kit from the RNAs. Gene expressions were measured by real-time polymerase chain reaction (PCR). Expression levels were quantitated according to the 2−ΔΔCT method.
Statistical analysis
SPSS 19.0 statistical program was used for statistical analysis. Student's t-test values are shown as mean ± standard deviation with differences between groups.
Results
Determination of DZNep concentration and effect of DZNep on cell viability
The concentration of DZNep in HepG2 cells was determined by MTT test. HepG2 cells were treated at different concentrations of DZNep (0.1, 0.5, 1, 2, 2.5, 5, 7.5, 10 μM). The IC50 value was determined as 5 μM, and this dose was used for further experiments (Fig. 1A). Then addionally, we evaluated the effect of the determined dose in a time-dependent manner. Therefore, HepG2 cells were treated with 5 μM DZNep for 24, 48, 72, 96, and 120 hours, respectively. DZNep reduced cell proliferation for 72 hours (Fig. 1B).

Effect of DZNep on colony formation in HepG2 cells
DZNep, an epi-drug, targeting specific enzymes that play a role in epigenetic regulation, has an anticancer effect against metastasis and proliferation (Fujiwara et al., 2014; Li et al., 2013). We next examined whether DZNep reduced colony formation in HepG2 cells. DZNep significantly reduced colony formation (p < 0.05) (Fig. 2).

HepG2 cells were treated with DZNep for 72 hours and after 9 days incubation, stained with Crystal Violet and counted. DZNep significantly reduced colony formation compared with the control group (*p < 0.05).
Effect of DZNep on apoptosis in HepG2 cells
Apoptosis evasion is present regardless of the cause or type in all cancer cells. Therefore, it is an important approach to stimulate the apoptosis mechanism. We evaluated the effect of DZNep on apoptosis in HepG2 cells. Therefore, accordingly it was demonstrated that DZNep induced total apoptosis at 8.5%, through Annexin-V-FITC/PI staining in HepG2 cells. However, there was no statistically significant difference in early and late apoptosis compared with the control group (p > 0.05; p < 0.05) (Fig. 3).

Detection of apoptosis was performed based on detection of PS on the surface of apoptotic cells and using a premixed reagent containing fluorescently labeled and measure by flow cytometry. There was no statistically significant difference early and late apoptosis, but DZNep statistically increased the total apoptosis compared with control group (p > 0.05; *p < 0.05). PS, phosphatidylserine.
Effect of DZNep on EZH2, EMT and ECM marker gene expressions
DZNep is known as an antitumor agent through EZH2 inhibition (Fujiwara et al., 2014; Li et al., 2013). Therefore, first, we wanted to prove whether DZNep truly inhibits EZH2 as shown by real-time PCR. We have shown that DZNep significantly decreased EZH2 gene expression level (p < 0.01) (Fig. 4A).
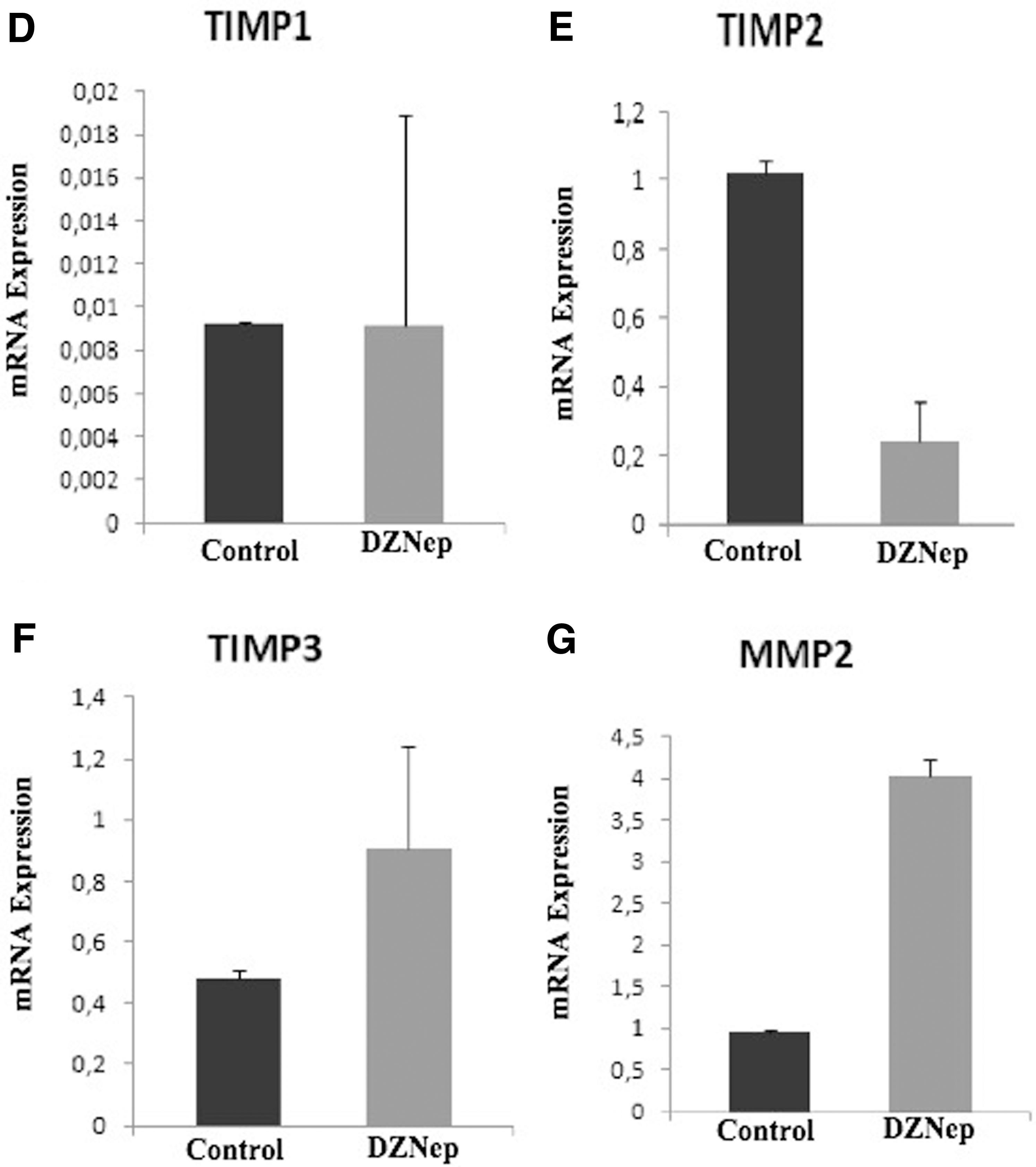
Then, we evaluated the effect of DZNep treatment on EMT in HepG2 cells by also real-time PCR. Interestingly; inhibition of EZH2 through epi-drugs is associated with poor prognosis, in contrast to what is known in various types of cancers. It has also been reported that epi-drugs increase the invasion and metastasis in cancer cells by activating the EMT pathway, which plays an important role in cancer (Cardenas et al., 2016).
DZNep decreased epithelial markers of E-cad and increased mesenchymal marker of Vim expression levels (respectively, p < 0.01; p < 0.01) (Fig. 4B, C).
Basal membrane and ECM degradation have been shown to be the main step of tumor invasion and metastasis mechanisms in new cancer studies. Malignant tumors use MMPs to overcome this ECM barrier (Chen et al., 2013). The level of MMP expression increases in many cancer cells, such as lung, breast, colon, stomach, and brain. MMP levels were associated with invasive and metastatic behavior in these tumors. MMP activity is regulated by TIMPs, which are specific tissue inhibitors for MMPs (Nagase, 1997).
Lastly, we evaluated the effect of DZNep treatment on ECM marker in HepG2 cells by real-time PCR.
There was no statistically significant difference in TIMP1 gene expression level. DZNep decreased ECM marker of TIMP2, increased MMP2 and TIMP3 expression levels (respectively, p > 0.05, p < 0.01, p < 0.01, p < 0.01) (Fig. 4D–G).
Discussion
Recent studies have shown that EZH2 plays a role as an oncogene, the overexpression causes tumor development, and has also effects on cell cycle and apoptosis (Girard et al., 2014; Wassef et al., 2016). It has been shown that EZH2 is overexpressed in a different type of solid tumors, such as prostate (Bachmann et al., 2006), breast (Kleer et al., 2003), colon (Katona et al., 2014), liver (Zhai et al., 2016), and hematological malignancies (Herviou et al., 2016; Wang et al., 2015). Various epi-drugs targeting specific enzymes that play a role in epigenetic regulation are being researched as important due to having an anticancer effect against HCC metastasis and proliferation (Han et al., 2018).
Therefore; in our study first, we investigated the effect of 72 hours of DZNep treatment on EZH2 gene expression, cell proliferation, colony formation, and apoptosis in the HepG2 cells. DZNep is known to be an S-adenosyl homocysteine hydrolase inhibitor (Fujiwara et al., 2014). DZNep inhibits the trimethylation of several lysine sites of histone H3, including lysine 27 (H3K27me3) of EZH2 (Fujiwara et al., 2014; Li et al., 2013).
Similar to other studies, we found that DZNep inhibited the EZH2 gene expression level, cell proliferation, and colony formation, at the same time induced apoptosis in HepG2 cells.
Hibino et al. (2014) have reported that DZNep-induced apoptosis, G0/G1 phase arrest, and inhibits cell proliferation in HepG2 cell line.
In another study on mice; HCC cells have been implanted into the mice and (Chiba et al., 2012) have observed that the cells' self-renewal capacity decreased with the tumor-initiating cells due to the inhibition of EZH2 through DZNep in HCC cells. Therefore, it has been suggested that the tumorigenic activities of these cells are dependent on EZH2. Accordingly, it has been reported that the inhibition of EZH2 by pharmacological intervention may be a promising therapeutic approach to target tumor-initiating HCC cells.
Au et al. (2013) have reported that inhibition of EZH2 through DZNep recover reverse activation of DLC1 expression isolated from HCC cells in different cancer cell lines. EZH2-mediated H3K27me3 epigenetic regulation of DLC1 is a common mechanism in human cancers. Also Au et al. (2013) have shown that DZNep treatment reduces the migration of HCC cells by disrupting the actin cytoskeleton binding and that DZNep may have a significant therapeutic effect in targeting cancer metastasis.
In another study, Pan et al. (2014) have shown that DZNep treatment induces apoptosis and G0/G1 phase arrest in gastric cancer cell lines. Studies are very insufficient on the effect of DZNep in HCC.
As a result; our findings, as in the cancer studies mentioned above, we observed the positive effects of DZNep treatment (reducing the proliferation of cancer cells, inducing apoptosis and reducing colony formation) in HCC.
In tissue repair and pathological processes, especially the origin of mesenchymal cells involved in tissue fibrosis, tumor invasion, and metastasis is not fully understood. However, emerging evidence suggests that EMT is an important effect in these cells (Kalluri and Weinberg, 2009). Epithelial cells change their phenotype during EMT processes and acquire metastatic and invasive features (Liu et al., 2014).
Second, we also investigated the effects of DZNep on genes (E-cad, Vim) that play a role in EMT pathway and genes (MMP2, TIMP1, TIMP2 and TIMP3) that play a role in ECM.
In our study, inhibition of EZH2 was given unexpected results on HCC. We found that DZNep decreased the epithelial marker E-cad gene expression level, as well as increased the mesenchymal marker Vim gene expression level in HepG2 cells. In various cancer research, another target of EZH2 is a critical epithelial marker E-cad gene for EMT and metastasis (Fujii and Ochiai, 2008; Wang et al., 2013).
In many studies, EZH2 has been reported to reduce the E-cad expression by interacting with the EMT transcription factor SNAIL (Peinado et al., 2004).
Since DZNep-mediated EZH2 inhibition reduced cell proliferation, we thought that the remaining cells gained a more aggressive cancer phenotype and that EMT was induced.
Cardenas et al. (2016) reported that the reaction of H3K27 me3 catalyzed by EZH2 is necessary for the permanency of the normal epithelial phenotype in ovarian cancer cells and also reported that EZH2 inhibition stimulates the EMT process by increasing the activation of various transcription factors especially, ZEB2.
GSK126 (GSK2816126A), an EZH2 methyltransferase inhibitor, increases the expression level of the Vim-causing cells to acquire a mesenchymal phenotype. Cardenas et al. (2016) have shown that by enzymatic inhibition of EZH2, both siRNA and GSK126 mediated cause EMT-like modification in ovarian cancer cells.
In cancer studies, various EMT-related transcription factors have been identified, such as transforming growth factor beta (TGF-β), Wnt, Snail/Slug, Twist, and Six1. It has been proven that EMT regulators are associated with poor prognosis and tumor aggressiveness in clinical studies (Jiang et al., 2013; Syed, 2016).
Cardenas et al. (2016) and Syed et al. (2016) have reported that the level of TGF-β expression increased in ovarian cancer, thus epithelial cells gained migratory and invasive features with the loss of polarity, and also the level of ZEB2 expression increased with the activation of TGF-β. ZEB2 is an important transcription factor and activated in the late period of EMT (Syed et al., 2016). DZNep represses EZH2-mediated H3K27me3, so the decrease of H3K27me3 may be directly related to the increase of ZEB2.
In another study, it has been reported that smooth muscle-22a (SM22a), also known as transgelin (TAGLN), is expressed in mesenchymal cells, including myofibroblasts and smooth muscle cells and is involved in EMT process (Tawfik et al., 2014). Maleszewska et al. (2015) have shown that the inhibition of EZH2 stimulates IL-1β and TGFβ2 in endothelial cells and that correlates with decreased H3K27me3 levels in the TAGLN proximal promoter. Accordingly, the result is, EZH2 has been shown to regulate the chromatin structure in the TAGLN promoter by trimethylation of H3K27.
EZH2 also has a main regulatory function in the control of processes, such as stem cell differentiation, cell proliferation, early embryogenesis, and inactivation of the X chromosome. It has been shown that GSK126-mediated EZH2 inhibition in myeloid cells leads to new myeloid differentiation of origin from primitive hematopoietic progenitor cells. At the same time, this study revealed a mechanism that disappointed the results of phase I clinical trial of GSK126 (Huang et al., 2019).
In our study, we found that DZNep increased the levels of MMP2 and TIMP3 expression, one of the extracellular regulators of EMT, and did not change the level of TIMP1 gene expression, and decreased the level of TIMP2 gene expression.
It was an unexpected situation for us that DZNep increased the level of MMP2 and decreased TIMP2 expression in cancer cells because MMP2 has been shown to play a promoter role in initiating the invasion and metastasis of cancer cells (Scheau et al., 2019). It has been reported that TIMP2 both inhibits active MMP2 and activates pro-MMP2 by activation of membrane-bound MMP14. However, TIMP2 has also been shown to play an endogenous inhibitor role in tumor development (Bourboulia et al., 2013).
Anne and Taina (2010) have analyzed the levels of serum MMP-2-TIMP-2-complex, MMP-9, TIMP-1, and TIMP-2 using ELISA method in cervical cancer patients. They have observed that the mean levels of serum TIMP-2 and MMP-2/TIMP-2 complex were higher in healthy individuals and a low level of TIMP 2 expression is associated with poor prognosis in cervical cancer.
Peeney et al. (2020) have investigated the effects of exogenous TIMP2 treatment on tumor development and metastasis in mouse models, which were phenotypically similar to human triple-negative breast cancer. As a result, TIMP2 treatment has been shown to suppress primary tumor development by more than 36%–50% and pulmonary metastasis 92%. In addition, lung tissue lysates have been taken in these mouse models. Peeney et al. (2020) have also reported that TIMP2 treatment disrupted PI3K signaling, which modulates proliferative and metastatic behavior along with WNT signals in the EMT pathway and reported that TIMP-2 has positive effects on suppression of triple-negative breast cancer growth and metastasis by modulating signal paths associated with EMT and metastatic growth.
In this study, despite DZNep treatment, low TIMP2 levels cause more active MMP2 or low TIMP2 production leads to less MMP2 inhibition. Therefore, we thought that this imbalance induces EMT that causes epithelial cells to synthesize excessive amounts of ECM proteins and contributing to the transformation of epithelial cells into mesenchymal cells.
In our study, we found that DZNep increased TIMP3 expression levels as opposed to TIMP2 expression.
It has been shown that TIMP3 expression levels are low in various cancers. Real-time PCR (RT-PCR) and western blotting analysis have shown that the level of TIMP-3 expression and protein expression is reduced in four HCC cell lines (BEL-7402, SMMC-7721, HepG2, and SK-HEP-1) and tissues compared with tissue and cell of healthy individual (Gu et al., 2014). In addition, decreased TIMP3 expression levels have shown to be negatively correlated with certain clinical pathology items, including portal vein invasion and lymph node metastasis. Also, researchers have been shown that TIMP3 suppresses in vivo tumor development and that liver metastasis is significantly reduced by TIMP3 transduction (Wang et al., 2010; Zhang et al., 2007).
Decreased TIMP3 expression levels may be related with the loss of ZEB1 in this study. According to these results, an increase of TIMP3 expression levels was evaluated positively in terms of preventing the EMT process in HepG2 cell line.
In our study, we found that DZNep did not change the TIMP1 expression level in HCC.
It has been reported that TIMP1 regulates intracellular signaling pathways for inhibition of apoptosis and activates the EMT process, causing changes in cell morphology in EMT (Angelo et al., 2014). TIMP-1 expression of HCC tissues is associated with advanced TNM stage, intrahepatic metastasis, portal vein invasion, and vasculature invasion. Experimental studies have shown that the level of TIMP1 expression is lower in normal human hepatocyte LO2 cells. After liver resection in HCC tissue, it has been observed that TIMP-1 expression is significantly associated with worse overall survival in HCC patients (Song et al., 2015).
We have observed that DZNep does not show any effect on TIMP1, where it acts mostly on TIMP2 and TIMP3 in HCC.
Interestingly, it has been reported that EZH2 has canonical roles on nonhistone protein methylation, such as JARID2 protein, a noncatalytic member of PRC2, and STAT3 pathway causing the induction of the EMT (Tange et al., 2014). Since EZH2 has a complex effect on cancer treatment, we have suggested that the effects of inhibition or activation should be investigated more comprehensively.
In this study, we observed a positive effect of DZNep, on account of the fact that DZNep induces apoptosis and TIMP3 expression level, and decrease in colony formation.
We think that epigenetic drugs would have complicated results because of affecting many substrates, such as decrease in the level of gene expression E-cad and TIMP2, and increase in the level of Vim and MMP2 expression.
As a result, epigenetic drugs have shown beneficial effects for the treatment of hematological malignancies, but some clinical studies have failed to show the effectiveness of these epigenetic drugs in treatment for HCC and other solid tumors (Lu et al., 2020).
Conclusion
There may be potential hazards in the use of demethylation agents in anticancer therapy due to their potential effects on activating a large number of silent genes, including genes involved in promoting tumor metastases.
We think that further studies are necessary to clarify this issue and should be attentive using DZNep in cancer treatment.
Footnotes
Acknowledgments
This work was done in the Betül-Ziya Eren Genome and Stem Cell Center and Department of Biochemistry Medical Faculty in Erciyes University, Kayseri/Turkey.
Author Contrıbutıons
G.B. and M.Ö. designed the study. E.K., G.B., and M.Ö. wrote the article. M.B., G.B., M.Ö., and H.A. collected and analyzed the data. All authors read and approved the final article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was supported by the Grand Erciyes University, Kayseri/Turkey (Grand no.: TYL-2017-7179).