
Abstract
Induced pluripotent stem (iPS) cells have been produced just for a few species among order Carnivora: snow leopard, Bengal tiger, serval, jaguar, cat, dog, ferret, and American mink. We applied the iPS cell derivation protocol to the ringed seal (Phoca hispida) fibroblasts. The resulting cell line had the expression of pluripotency marker gene Rex1. Differentiation in embryoid body-like structures allowed us to register expression of AFP, endoderm marker, and Cdx2, trophectoderm marker, but not neuronal (ectoderm) markers. The cells readily differentiated into adipocytes and osteocytes, mesoderm cell types of origin. Transcriptome analysis allowed us to conclude that the cell line does not resemble human pluripotent cells, and, therefore, most probably is not pluripotent. Thus, we produced ringed seal multipotent stem cell line capable of differentiation into adipocytes and osteocytes.
Introduction
Order Carnivora consists of two suborders: Caniformia and Feliformia. It includes domestic animals that are used for disease modeling; fur-bearing animals, such as mink, silver and arctic foxes, marine mammals, and other species. Derivation of induced pluripotent stem (iPS) cells could allow insights into pluripotency and embryonic development of these species, as well as the development of new disease models. Currently, pluripotent cells were produced from several Carnivora species: dog (Baird et al., 2015; Koh et al., 2012; Lee et al., 2011; Luo et al., 2011; Shimada et al., 2010; Tsukamoto et al., 2018; Vaags et al., 2009; Whitworth et al., 2012), snow leopard (Verma et al., 2012), Bengal tiger, serval, jaguar, and cat (Dutton et al., 2019; Gómez et al., 2010; Verma et al., 2013), ferret (Gao et al., 2020), and American mink (Menzorov et al., 2015).
Marine mammals represent the basal Carnivora group. Many pinniped species are considered endangered. The tissue and cell samples are needed to study their biology. Also, as their embryos are essentially unavailable, iPS cells would expand our knowledge of their embryonic development and differentiation. The ringed seal is one of the species that may suffer from global climate change, as it needs sea ice to live. We attempted to generate ringed seal (Phoca hispida) iPS cells. We present a case study of a multipotent stem cell-like clone that expressed pluripotency and mesenchymal stem cell (MSC) marker Rex1 and was able to differentiate them into mesoderm cell types, adipocytes and osteocytes.
Materials and Methods
Production of ringed seal fibroblasts
Primary fibroblasts of the ringed seal were obtained from lung necropsy. Tissue samples from wild female were collected during aboriginal quota sealing in the coastal waters of the Bering Sea (Mechigmen bay, Chukotka Autonomous Okrug, Russia). To establish primary fibroblast cell culture, we used a conventional technique (Stanyon and Galleni, 1991) with modifications. Lung necropsy was delivered to the laboratory in the biopsy tissue medium: a modification of minimum essential medium (a-MEM) with 15% fetal bovine serum (FBS), amphotericin β (6 μg/mL), streptomycin (300 μg/mL), and penicillin (300 μg/mL) (Thermo Fisher Scientific, Waltham, MA).
The sample was washed twice in a Dulbecco's Modified Eagle Medium (DMEM) with the same antibiotics and antimycotic, transferred to a small Petri dish, and minced with scissors. The fragmented sample was resuspended in a 1 mg/mL solution of Type I collagenase (Thermo Fisher Scientific) and 1 mg/mL Type I-S hyaluronidase from bovine testes (Sigma-Aldrich, Burlington, MA) in the biopsy tissue medium with 10% FBS. After overnight culture at 37°C, cells were centrifuged and the residue resuspended in the fibroblast culture medium. The fibroblast culture medium consisted of a-MEM supplemented with 15% FBS, 1 × MEM nonessential amino acids solution, 1 × GlutaMAX supplement, and 1 × penicillin/streptomycin (Thermo Fisher Scientific). Cell culture was performed at 37°C and 5% CO2.
Production of ringed seal multipotent stem cell line
To reprogram the ringed seal fibroblasts, we used lentiviral vectors LeGO (www.lentigo-vectors.de/vectors.htm) with EGFP and human reprogramming transcription factors: OCT4, SOX2, C-MYC, and KLF4, courtesy of Dr. Sergei L. Kiselev, Moscow. We used the previously published protocol (Menzorov et al., 2015) with minor modifications. Lentiviruses were produced in the Phoenix cell line using Lipofectamine 3000 (Thermo Fisher Scientific). The multiplicity of infection was estimated as 5.1 using EGFP lentiviral vector (Beklemisheva and Menzorov, 2018). Fibroblasts at passage 4 (3 × 105 cells, 30 × 103 cells/cm2) plated the day before were transduced with viruses containing four reprogramming transcription factors: 50% virus supernatant, 50% fibroblast culture medium without antibiotics with heat-inactivated FBS, and 10 μg/mL Polybrene.
Transduction was performed for two consecutive days. Cells were passaged onto a 6-cm cell culture dish coated with 0.1% gelatin on feeder cells on day 5. We used mitomycin C-treated 13.5 dpc fibroblasts of the CD-1 mouse strain as feeder cells (Hogan et al., 1994). From day 6, we used iPS cell culture medium: a-MEM supplemented with 20% ES (embryonic stem) cell qualified FBS, 1 × MEM nonessential amino acids solution, 1 × GlutaMAX supplement, 0.1 mM 2-mercaptoethanol, 1 × penicillin/streptomycin, and 10 ng/mL basic fibroblast growth factor (bFGF) recombinant human protein (Thermo Fisher Scientific).
From day 6 to 12 the medium was changed once in 2 days with the addition of 1 mM valproic acid (Sigma-Aldrich). On day 19 we observed flat colonies with nonfibroblast morphology. On day 27 colonies morphologically distinct from fibroblast colonies were picked up and expanded on the feeder. We were not able to find colonies with “naïve” mouse, “primed” human, or American mink ES cell morphology, so the selection based on morphology was subjective, as the ringed seal pluripotent stem cell morphology is unknown. The passage was performed with 0.25% Trypsin-EDTA (Thermo Fisher Scientific). All cell cultures were maintained at 37°C and 5% CO2.
Multipotent stem cell derivation was performed at the Collective Center of ICG SB RAS “Collection of Pluripotent Human and Mammalian Cell Cultures for Biological and Biomedical Research” (http://ckp.icgen.ru/cells/; www.biores.cytogen.ru/icg_sb_ras_cell/).
All animal studies were undertaken with prior approval from the Ethics Committee on Animal and Human Research of the Institute of Molecular and Cellular Biology SB RAS, Russia (protocol No. 01/20 of February 11, 2020).
Cytogenetic analysis
Cytogenetic analysis for fibroblasts was carried out on passage 7 and for a multipotent stem cell line on passage 9. The preparation of metaphase chromosomes from fibroblasts was performed as previously described (Graphodatsky et al., 2000, 2001; Yang et al., 1999). GTG-banding of metaphase chromosomes was done according to a previously published protocol (Seabright, 1971). For each cell line, averages of 50 conventionally stained by Giemsa metaphase plates were analyzed. Digital images were captured using the VideoTest system (Zenit, St. Petersburg, Russia) with a charge-coupled device (CCD) camera (Jenoptik, Jena, Germany) mounted on a Zeiss microscope, Axioscope 2 (Zeiss, Oberkochen, Germany). Metaphase spread images were edited in Corel Paint Shop Pro Photo X2 (Corel, Ottawa, Canada). Chromosomes of the ringed seal (P. hispida) were arranged according to the current nomenclature (Graphodatsky et al., 2020).
Multipotent stem cell differentiation
Differentiation into embryoid body-like (EB-like) structures was performed in EB differentiation medium, the same composition as iPS cell culture medium, but with 20% FBS instead of ES = qualified FBS and without bFGF. Cells were passaged into 1% agarose-coated cell culture plates to prevent attachment. The medium was changed every second day for 31 days. On day 5, part of EBs were plated onto 0.1% gelatin-coated cell culture plates for osteocyte and adipocyte differentiation. Osteocyte differentiation was carried out in EB differentiation medium; adipocyte differentiation in a similar adipocyte differentiation medium supplemented with 10% knockout serum replacement (KSR) (Thermo Fisher Scientific) instead of FBS from day 6.
Cytochemical staining
Cells were fixed by 4% paraformaldehyde for 20 min and washed with phosphate-buffered saline (PBS). Adipocytes were stained with 0.7% lipid detection stain Sudan Black B in propylene glycol for 20 min, washed twice with 0.85% propylene glycol, and washed multiple times with PBS. Osteocyte calcification was shown by specific staining. Osteocytes were stained with 2% Alizarin Red S (Sigma-Aldrich) solution in water, pH = 4.23, for an hour and washed multiple times with PBS. Staining was analyzed on Zeiss Observer.Z1 fluorescent microscope with AxioCam HRm 3 CCD-camera (Zeiss). Digital images were analyzed using the ZEN 2 starter (Zeiss) software.
DNA isolation
Genomic DNA was isolated from cells using a polymerase chain reaction (PCR) buffer with nonionic detergents, which was adapted from a protocol from PerkinElmer Cetus (Higuchi, 1989). We used the DNA for the Mycoplasma contamination testing. Both ringed seal fibroblasts and iPHIS1 cells were negative for Mycoplasma contamination.
RNA isolation and cDNA synthesis
RNA was isolated using the Aurum Total RNA Mini Kit (Bio-Rad, Hercules, CA) according to the manufacturer's recommendations. We isolated RNA from one million cells for the subsequent bulk RNA-seq transcriptome (one biological replicate) or reverse transcription-PCR (RT-PCR) analysis. RNA was treated with DNaseI from the kit during the isolation. For RT-PCR analysis we additionally applied DNaseI (Fermentas, Waltham, MA), then 0.4 micrograms of total RNA was used for cDNA synthesis by the First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific).
Primer design and PCR
We used previously published primer sequences and Primer-BLAST software (Ye et al., 2012) to design primers for mink Rex1 and canine Cdx2 (Table 1). PCR was performed using BioMaster HS-Taq PCR-Color (2 × ) (Biolabmix, Russia) in 10 μL reaction volume.
Primers for Reverse Transcription-PCR (RT-PCR)
BLAST, Basic Local Alignment Search Tool.
De novo transcriptome assembly and dataset annotation
We performed nonstranded and polyA-selected mRNA library preparation and PE100 sequencing on DNBSEQ at BGI (People's Republic of China) (Drmanac et al., 2010; Huang et al., 2017). The number of reads was 62,390,350, Q20 rate—95.77%, and GC rate—50.78%. The raw sequence data are available in NCBI BioProject repository, accession number: PRJNA718133, link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA718133.
RNA-seq read quality was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The RNA was isolated from cell culture containing mouse feeder cells. To exclude reads originating from feeder transcripts we aligned reads to the mouse genome using bowtie2 (Langmead and Salzberg, 2012) with default parameters. All aligned reads were excluded from the following analysis. De novo transcriptome assembly was done with Trinity (v. 2.11.0) using default settings (Grabherr et al., 2011; Haas et al., 2013).
Coding regions of the assembled transcripts were predicted using TransDecoder (v. 5.5.0) with default settings (Haas et al., 2013). We used the Basic Local Alignment Search Tool (BLAST) and the Swiss-Prot database to assign functional annotations (Altschul et al., 1990; Camacho et al., 2009). We assessed the quality of transcriptome assembly using BUSCO (Seppey et al., 2019) and Trinity scripts. The following filtration was done using homemade Python scripts. We used StringTie for generating transcript quantifications with default options (Pertea et al., 2015). RNA-seq data analysis was performed using https://usegalaxy.org/ server (Afgan et al., 2018) and Computational Cluster of the Novosibirsk State University (Russia).
Analysis of endo- and exogenous expression of OCT4, KLF4, SOX2, and c-MYC genes
We extracted all ringed seal transcripts encoding OCT4, KLF4, SOX2, and c-MYC using BLAST against the Swiss-Prot database. For this analysis, we used transcriptome assembly obtained from unfiltered reads (i.e., possibly containing mouse feeder transcripts), to allow capturing of transcripts conserved between mouse, human, and ringed seal. We found that each of the target genes was represented by multiple Trinity transcripts. We manually annotated all obtained transcripts using BLAST nucleotide alignment and BLAST protein alignment against NCBI nucleotide or protein collections. For three genes (except Sox2), we found that some transcripts showed better alignment to Carnivora orthologs, whereas other transcripts had better alignment to primate orthologs. These results were consistent between nucleotide and protein alignments and therefore allowed us to classify transcripts by organism of origin.
To qualitatively estimate expression levels of the target genes, we used bowtie2 (Langmead and Salzberg, 2012) to align reads to assembled transcripts of ringed seal and human orthologs. Next, the reads were classified as those aligned specifically to human reference, specifically to ringed seal reference, and to both references. We performed alignment and classification, both for the raw reads and reads filtered against mouse references (see above). The obtained counts for each class were used to plot Figures 2C and D.
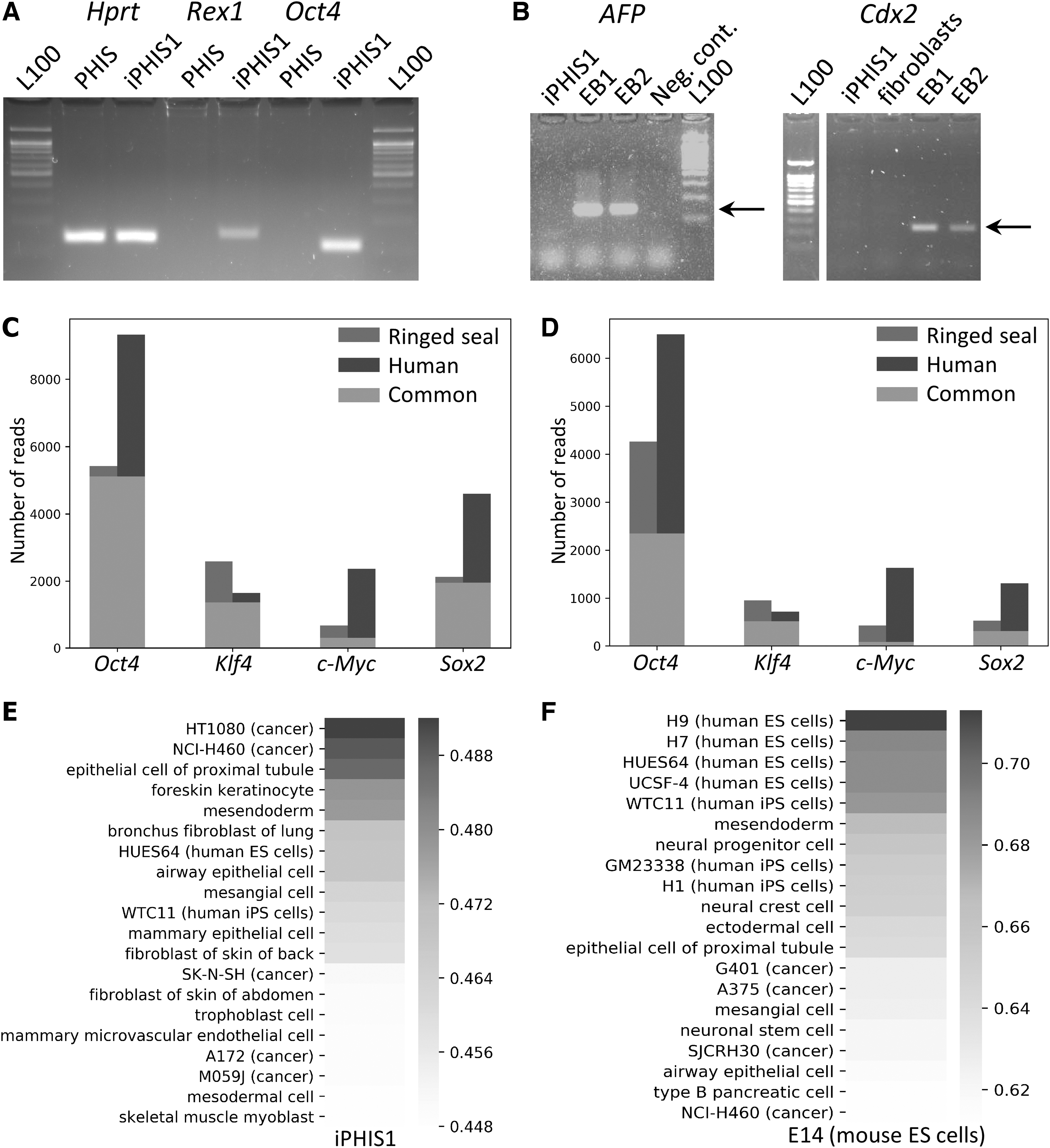
Gene expression in iPHIS1 and transcriptome analysis of ringed seal induced multipotent stem cells.
Genome-wide comparison of gene expression between cell types and species
We obtained gene expression quantification files from ENCODE using batch download with a filter (organism: human; assay: RNA-seq; file format: tsv). To cover more cell types, we included both total- and poly(A)-enriched RNA-seq datasets in analysis. All files were processed using homemade Python scripts to obtain FPKM values. We filtered out genes that do not have one-to-one orthologs among mouse, human, and ringed seal. For the remaining genes, we only kept the top 10% of genes that showed the highest FPKM standard deviation values across samples. The resulting gene expression matrix (1843 genes) was used to compute Spearman's R correlation coefficient between each pair of samples.
Results
Multipotent stem cell derivation and differentiation
The experiment outline is shown in Figure 1A. We aimed to produce iPS cells from primary ringed seal fibroblasts and used the following human reprogramming transcription factors: OCT4, SOX2, C-MYC, and KLF4. On day 27 after transduction, there were colonies with morphology distinct from fibroblasts. Two colonies were picked up and expanded (Fig. 1B). One of them, iPHIS1, gave rise to cells that grew in a monolayer (Fig. 1C, D), the other had fibroblast morphology after the passage and was discarded. Overgrown culture formed “bubbles” (Fig. 1E) that later detached and floated as cyst-like structures resembling canine embryos (Hayes et al., 2008). We cultured cells up to passage 15 with no signs of proliferation decline.
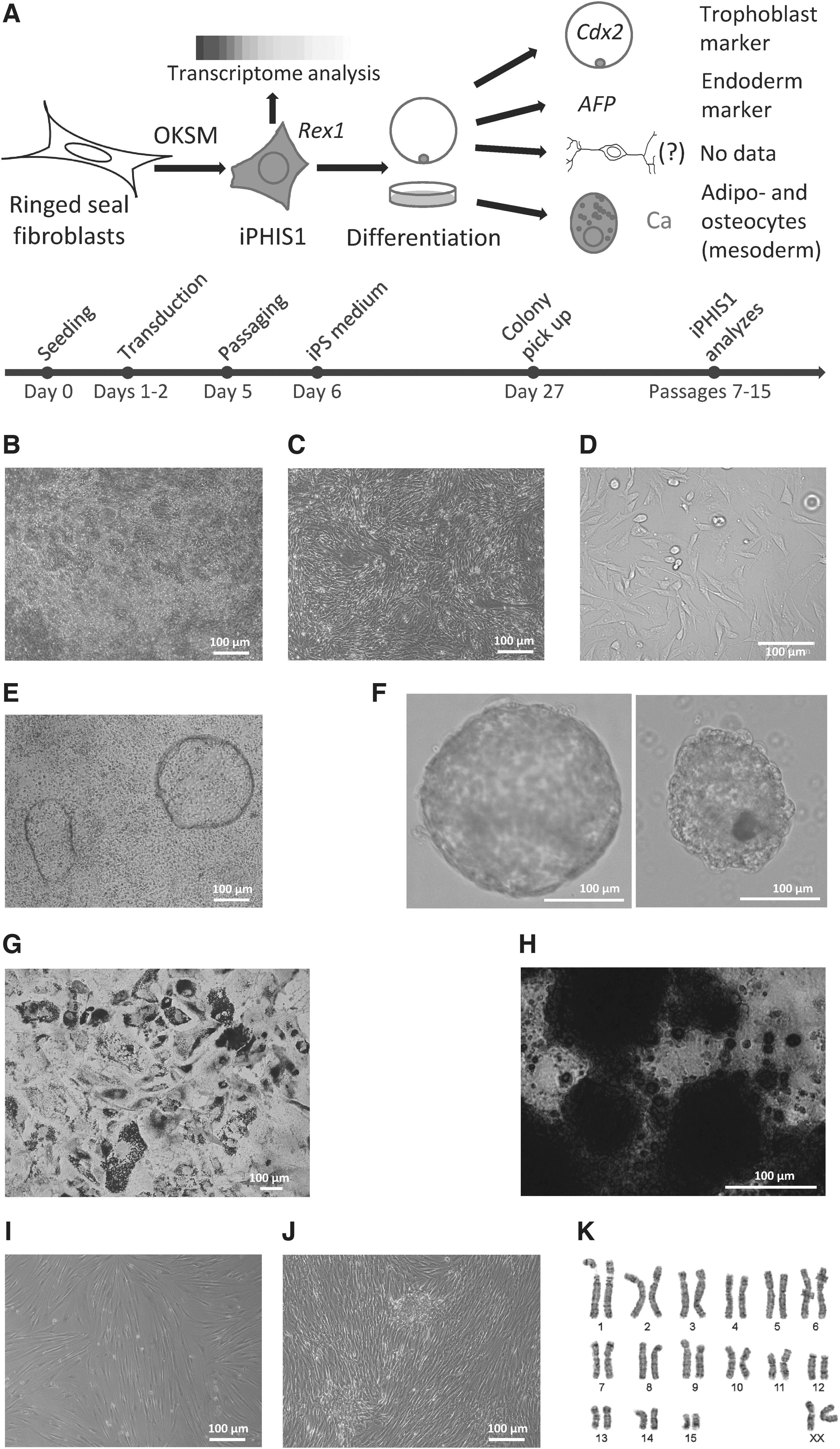
Derivation, colony morphology, differentiation, and karyotype of ringed seal induced multipotent stem cells.
We attempted to differentiate iPHIS1 cells at passage 7 into derivatives of the three germ layers. Solid and cyst-like EB-like structures were formed after passage to nonadhesive culture plates (Fig. 1F). Their gene expression analysis is described below. Adipogenic and osteogenic differentiation was successfully performed in the adipogenic and EB differentiation media, respectively (Fig. 1G, H). We used KSR-based neuronal differentiation medium developed for canine ES cells (Hayes et al., 2008) in an attempt to differentiate cells into neurons, instead cells differentiated into adipocytes. Interestingly, there was some calcification in the adipocyte differentiation medium (data not shown). We used primary ringed seal fibroblasts at passage 8 as a negative control for adipogenic and osteogenic differentiation using the same protocol. There were no adipocytes and calcification (Fig. 1I, J).
Cytogenetic analysis
We performed the cytogenetic analysis of the ringed seal (P. hispida) primary fibroblasts and iPHIS1 cells at passages 7 and 9, correspondingly. Fibroblasts had 32 chromosomes, typical for this species (Árnason, 1974), XX (n = 50) (Fig. 1K) with 15.7% polyploid cells. The iPHIS1 cells (n = 50) had 32 (n = 47), 33 (n = 2), and 35 (n = 1) chromosomes, with 5% polyploid cells. We had not revealed a difference in the pattern of GTG-banding between karyotypes of the primary fibroblasts and iPHIS1 cells.
Gene expression pattern
First, we applied RT-PCR analysis of gene expression at passage 12. Primers were 95%-100% homologous to P. hispida Hprt1 and Oct4 genes or a close species, P. vitulina, Rex1, AFP, and Cdx2 genes with a mismatch in the middle. We were able to show the expression of one of the key pluripotency markers Rex1 (Zfp42) as well as Oct4 in iPHIS1 cells by RT-PCR (Fig. 2A). Analysis of EB-like structures revealed the expression of AFP, endoderm marker, and Cdx2, trophoblast marker (Fig. 2B).
We next employed transcriptome sequencing to profile genome-wide gene expression in iPHIS1 cells at passage 15, one replicate. To avoid contamination originating from mouse feeder cell transcripts, we filtered out all reads aligned to the mouse genome (8.5% of reads) and subjected the remaining dataset to de novo transcriptome assembly. This resulted in a draft assembly composed of 170,758 genes (209,671 transcripts) (https://dx-doi-org.web.bisu.edu.cn/10.17632/5mkvk5yc4w.1). BUSCO ortholog analysis showed that 67% out of the Mammalia BUSCO database were present in this draft assembly, confirming its high quality.
We then used the Swiss-Prot protein database to annotate obtained Baikal seal genes, which allowed us to find orthologs for 31,194, containing 12,228 unique gene names (https://dx-doi-org.web.bisu.edu.cn/10.17632/5mkvk5yc4w.1).
We used the obtained transcriptomic data to profile the expression of some marker genes in iPHIS1 cells (https://dx-doi-org.web.bisu.edu.cn/10.17632/5mkvk5yc4w.1). We confirmed a relatively high level of Rex1 expression. There was no Nanog pluripotency marker in the Trinity transcripts. We had manually aligned reads to P. vitulina Nanog and were able to find just 44 reads. American mink ES and iPS have a low level of Nanog expression compared with mouse and human pluripotent stem cells (Menzorov et al., 2015). Some percentage of cultured human MSCs also express Nanog (Pierantozzi et al., 2010). We concluded that iPHIS1 cells express Nanog at a low level.
As we introduced human OCT4, KLF4, SOX2, and c-MYC genes to reprogram fibroblasts into iPS cells, we decided to check whether those genes are expressed or silenced. Lentiviral transgenes are silenced in pluripotent stem cells; thus, their silencing would suggest passing through pluripotency. We performed an analysis of endo- and exogenous expression of their transcripts and found expression of both human transgenes and ringed seal orthologs of Oct4 (Pou5f1), Klf4, and c-Myc (Fig. 2C). We noted that filtering reads that align to mouse genome had not changed these results, suggesting that Klf4 and c-Myc expression does not originate from mouse feeder cells (Fig. 2C, D). As for Sox2, we found that Trinity failed to assemble a ringed seal ortholog. We used P. vitulina Sox2 reference to align P. hispida reads. We have found reads from both endo- and exogenous transcripts for all four reprogramming factors. Thus, there were no signs of transgene silencing.
Next, we performed a genome-wide comparison of expression patterns observed in iPHIS1 cells and various human cell types with added mouse ES cell line E14. For this analysis, we used all available human RNA-seq data from ENCODE, 288 samples. We accessed the similarity of gene expression profiles using Spearman's correlation coefficient (Fig. 2E). To test the applicability of this approach, we performed the same analysis comparing mouse ES cell line E14 transcriptome data with the same human cell lines and tissues (Fig. 2F). As expected, mouse ES cells showed the highest similarity to human pluripotent cells. In contrast, iPHIS1 expression shows the highest correlation with several cancer cell lines, as well as with foreskin keratinocytes, but not with pluripotent cells. These data argue against the establishment of pluripotency in iPHIS1 cells.
Discussion
Transcription factors, Oct4, Sox2, c-Myc, and Klf4, were used to generate iPS cells from a variety of Carnivora species. We decided to produce iPS cells from the ringed seal (P. hispida), the basal representative of the suborder Caniformia. Only one cell line expressed pluripotency marker genes Rex1 and Oct4. The iPHIS1 was diploid with a small proportion of tetraploid cells; it is normal for primary fibroblasts of various pinniped species (Árnason, 1974; Beklemisheva et al., 2020) as well as for pluripotent stem cells of different species. Its morphology differed from mouse, human, and mink iPS cells (Fig. 1C, D). Similar to American mink iPS cells, overgrown cell culture formed “bubbles” (Fig. 1E) that later unfastened and floated as cysts.
We differentiated iPHIS1 cells into EB-like structures to analyze the differentiation potential (Fig. 1F). Cyst-like EBs resembled canine embryos (Hayes et al., 2008), expressed endoderm marker AFP and trophoblast marker Cdx2 (Fig. 2B). EBs plated on gelatin efficiently differentiated into mesoderm derivatives: adipocytes and osteocytes. We were not able to show ectoderm differentiation, as Tubb3 marker gene was expressed in both iPHIS1 and EBs (data not shown). The presence of Tubb3 transcript in undifferentiated pluripotent stem cells is in accordance with our previous data on mouse ES cells (Menzorov et al., 2019). The ability to differentiate into adipocytes and osteocytes allows us to presume that iPHIS1 cells are at least multipotent.
The expression of a trophoblast marker upon differentiation, Cdx2, was rather unexpected, although trophoblast differentiation was shown for human and mouse primed pluripotent stem cells (Kojima et al., 2014; Xu et al., 2002) and canine iPS cells (Luo et al., 2011; Wilcox et al., 2009). If iPHIS1 cells were pluripotent, differentiation into trophoblast would suggest a primed pluripotency state.
We used bFGF to derive and propagate the iPHIS1 cell line. Colonies with prospective morphology were not formed in the media supplemented with leukemia inhibitory factor (LIF) or combination of LIF and bFGF. Canine pluripotent stem cells were produced with LIF (Baird et al., 2015; Lee et al., 2011; Luo et al., 2011; Vaags et al., 2009) or LIF and bFGF (Koh et al., 2012; Shimada et al., 2010; Tsukamoto et al., 2018; Whitworth et al., 2012). Also, cells were later cultured in the presence of LIF only (Whitworth et al., 2012). Other Carnivora species pluripotent cells include snow leopard (Verma et al., 2012), Bengal tiger, serval, jaguar, and cat (Dutton et al., 2019; Verma et al., 2013) cultured with LIF; cat cultured with LIF and bFGF (Gómez et al., 2010), and ferret cultured with bFGF (Gao et al., 2020).
Interestingly, cat iPS cells required species-specific feline LIF (Dutton et al., 2019), thus species-specific LIF or bFGF may be beneficial. We generated American mink iPS cells without LIF of bFGF supplementation (Menzorov et al., 2015), although inactivated mouse embryonic fibroblasts secrete both growth factors. Different requirements of the iPS cell culture of various species indicate that different signaling pathways are activated. It leads to different pluripotency states, naïve, primed, or other, as additional distinct pluripotency states had been described recently. More high-quality transcriptome data may facilitate the distinction between various pluripotency states in different species.
Pluripotent stem cells of different pluripotency states also have distinctive morphology. For instance, American mink pluripotent stem cells form colonies unlike mouse and human naïve and primed cells (Menzorov et al., 2015). Thus, morphology and expression of several marker genes are not enough to determine whether cells are pluripotent or not.
Transcriptome analysis of iPHIS1 cells revealed that their expression pattern resembled some human cancer cell lines, epithelial cells, keratinocytes, and mesodermal cells (Fig. 2E). Correlation of expression pattern of iPHIS1 and human pluripotent cells was slightly lower, although within the top quartile of all correlation coefficient values observed. Thus, transcriptome analysis allows concluding that iPHIS1 cells do not resemble human ES cells, as pluripotent stem cells were not among the closest by correlation. At the same time, this analysis applied for mouse ES cells was able to place mouse pluripotent cells close to human ones, thus this approach gives meaningful results.
Our data allow presuming that iPHIS1 cells are not pluripotent. The question remains, whether this cell line was produced by fibroblast reprogramming. If there was a pluripotent state, transgene silencing would be expected. Retroviral transgene silencing in pluripotent stem cells is a well-known phenomenon (Maherali et al., 2007; Okita et al., 2007; Wernig et al., 2007). The transcriptome data revealed expression of Oct4, Klf4, and c-Myc human transgenes comparable with their endogenous counterparts. As for Sox2, the endogenous expression was lower compared with the transgene. There are three main explanations.
First, iPHIS1 cells were reprogrammed in an intermediate pluripotency state and/or to iPS cells and subsequently differentiated into multipotent stem cells. The pluripotency state stage was short and not enough to silence the lentiviral transgenes.
Second, we directly reprogrammed ringed seal fibroblasts to multipotency. It was recently shown that Oct4, Klf4, Sox2, and c-Myc can reprogram somatic cells to a variety of cell types, including stromal ones (Schiebinger et al., 2019). Direct reprogramming could have been achieved by overexpression of OCT4 alone. CD34+ blood cells were successfully reprogrammed into MSCs by OCT4 overexpression and CHIR99021 treatment (Meng et al., 2013).
In addition, human dermal fibroblasts were chemically reprogrammed into MSCs (Lai et al., 2017), the authors hypothesize that the effect of the chemical cocktail was similar to OCT4 overexpression. The expression of endogenous and exogenous Oct4 was higher than for other reprogramming factors. Third, there is a possibility that a MSC in a fibroblast population gave rise to iPHIS1, probably even without reprogramming. It would explain the adipogenic and osteogenic differentiation of iPHIS1, as well as Oct4 and Rex1 expression. Rex1 and Oct4 are expressed in human MSCs (Bhandari et al., 2010; Han et al., 2014). As for AFP and Cdx2 gene expression (endoderm and trophoblast markers, respectively), their expression may be a culture artefact or a property of MSCs in given culture conditions, that is, during EB-like differentiation.
Most probably, iPHIS1 cells are MSCs. Based on our data we cannot prove it, as some other cell types can differentiate into adipocytes and osteocytes, such as pericytes. We noted that human MSCs are not among the top 20 cell types that showed the highest correlation of the gene expression levels. If iPHIS1 cells are MSCs, there are several possible explanations for this result: (1) interspecific differences between the ringed seal and human MSCs; (2) high heterogeneity between MSC populations; (3) expression of the human reprogramming factors that may “shift” expression profile.
We produced only one ringed seal multipotent stem cell line, experiments for iPS сell derivation were not successful. Suboptimal culture conditions might be the main reason. ES cell qualified FBS batch are only tested for mouse ES cell growth support. Also, growth factor origin may be important. We used human bFGF, and for cat iPS cells, only species-specific feline LIF was able to support pluripotency (Dutton et al., 2019). Growth factor concentration is an important factor as well. Ferret iPS cells were derived from human bFGF, but in a 10 × concentration compared with human iPS cells, and this study (Gao et al., 2020). Different researchers used small molecules to enhance iPS cell derivation efficiency.
In our experience, 2i inhibitors (PD0325901 and CHIR99021) and TGF-β antagonist A83-01 caused substantial fibroblast death, thus we were not able to use them. We applied valproic acid, histone deacetylase inhibitor, to facilitate reprogramming (Huangfu et al., 2008). Another way to increase efficiency is to use different transgenes and/or delivery vectors. We have recently shown that Sendai virus-based vector transgene expression level is superior to lentiviruses (Beklemisheva and Menzorov, 2018). Also, another set of reprogramming factors was successfully used to reprogram not only human, but ferret fibroblasts to pluripotency, OCT4, SOX2, KLF4, L-MYC, LIN28A, mp53DD, and 160 oriP/EBNA-1 (Gao et al., 2020).
Thus, we produced a ringed seal multipotent stem cell clone, capable of differentiation into mesoderm cell types, adipocytes, and osteocytes. We were able to show expression of endoderm (AFP) and trophoblast (Cdx2) markers after differentiation, but not ectodermal. Transcriptome analysis suggests that iPHIS1 cells do not resemble human or mouse pluripotent stem cells. Their differentiation profile suggests MSC identity, although we do not have sufficient data to prove it by transcriptome analysis. The iPHIS1 could be used as a model cell line for ringed seal adipogenesis and osteogenesis studies.
Availability of Data and Material
The raw sequence data are available in NCBI BioProject repository, accession number: PRJNA718133, link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA718133. The transcriptome analysis data are available in Mendeley Data repository: https://dx-doi-org.web.bisu.edu.cn/10.17632/5mkvk5yc4w.1
Ethics Approval
All animal studies were undertaken with prior approval from the Ethics Committee on Animal and Human Research of the Institute of Molecular and Cellular Biology SB RAS, Russia (protocol No. 01/20 of February 11, 2020).
Footnotes
Authors' Contributions
V.R.B. obtained ringed seal fibroblasts and performed cytogenetic analysis. A.G.M. produced and differentiated multipotent stem cells, performed RT-PCR gene expression analysis. P.S.B. performed transcriptome analysis. V.S.F. supervised transcriptome analysis. A.G.M. carried out interpretations of the data and project coordination. A.G.M. did most of the writing with contributions from V.R.B., P.S.B., and V.S.F. All authors read and approved the final article.
Acknowledgments
Authors are thankful to Michael Zelensky, Alexey Ottoj, and the Community of the Chukotka Autonomous Region indigenous “Lorino” (Russian Federation) for assistance in the ringed seal tissue sample collection. The authors gratefully acknowledge the primary fibroblast cell culture of P. hispida provided by the “Molecular and Cellular Biology” core facility of the Institute of Molecular and Cellular Biology SB RAS (project 0310-2018-0011).
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The reported study was funded by RFBR, project number 20-04-00369, and the Ministry of Education and Science of the Russian Federation, state project 0259-2021-0016.