
Abstract
Transdifferentiation means mature cell conversion into other mature cells. Ethical issues, epigenetic failure, or teratoma development are found in cellular reprogramming strategies. Thus, new methods are needed. This study aimed to develop a new novel formula of chemical molecules and growth factors that differentiate skin fibroblasts into insulin-producing cells (IPCs). Newborn mice fibroblasts differentiated using four induction methods into IPCs to search for the best method. Fibroblasts, stem cells, and pancreatic markers were identified using an immunocytochemistry (ICC) assay. Insulin was measured using ELISA and dithizone (DTZ) assays. The skin fibroblasts were induced successfully into IPCs. The best method to obtain IPCs was indicated by measuring insulin concentration in differentiated cell supernatant from all induced cells by the four methods. The protein expression of the pancreatic markers of induced cells increased with time, as indicated by the ICC assay. OCT3/4 increased on day 9, after which the expression tended to decrease. DTZ-positive clusters were observed on day 16. Secreted insulin of differentiated cells was injected in streptozotocin-induced diabetic mice, which decreased blood glucose levels after injection. This study indicated an efficient new chemical method for transdifferentiating skin fibroblasts into functional IPCs, which is a promising method for diabetes mellitus therapy.
Introduction
Cell therapy is a procedure used to administer, graft, or viably implant cells into an individual to accomplish a therapeutic act (Chari et al., 2018). Beta cells are large cells that store and release insulin in Langerhans' pancreatic islets to maintain glucose homeostasis (Fu et al., 2013). When the role of these cells is disturbed, this may cause type 1 or type 2 diabetes (American Diabetes Association, 2020). Diabetes is among the common and threatening diseases worldwide because other organs and tissues can be affected when untreated, leading to more serious conditions such as retinopathy, neuropathy, and angiopathy (Zimmet et al., 2016).
There have been attempts to treat and change the destruction of beta cells used for cell therapy, such as producing beta cells from various sources of stem cells. Stem cells are renewable multipotent and pluripotent cell sources that differentiate into different cell types, such as beta cells (Chen et al., 2020). Currently, β cell therapies focus on cells from different types of stem cells, including adult stem cells, embryonic stem cells (ESCs), and induced pluripotent stem cells (IPSCs) (Jun, 2010).
Many studies and approaches have used different sources of stem cells to produce beta cell-like cells or cells that secrete insulin effectively both in vitro and in vivo (Kroon et al., 2008). Despite the major and significant advantages of stem cells and their use in various fields, including cell therapy, they have some disadvantages, including the difficulty in isolating some of them. Moreover, limiting the number of ESCs tends to lead to teratoma after transplantation. Other disadvantages include ethical and religious dilemmas and immune rejection (Ghoneim et al., 2020). In addition, stem cells of adult origin, such as mesenchymal stem cells (MSCs), often need chronic and administrative care, and they are therapeutically ineffective and temporary (Takahashi and Yamanaka, 2006).
To address some of these obstacles and limitations, pluripotent stem cell development using nonpluripotent resources is now described as IPSCs. Somatic cells can be reprogrammed to develop pluripotent stem cells, and they can be divided into various cell types of IPSCs, which is accomplished by the direct expression of transcriptional factors (TFs), or Yamanaka factors, such as OCT4, SOX2, c-MYC, and KLF4. In 2006, IPSCs were discovered to open new possibilities for creating alternative cell-based therapy (Zhou and Zeng, 2013). TFs are introduced into cells by integrative and nonintegrative viral delivery methods. Episomal-, protein-, and chemical-based deliveries are all safe and secure (Nishimura et al., 2017). IPSCs and ESCs are similar to those from which pluripotent and self-renewable cells have been commonly used to produce in vitro and in vivo insulin-producing cells (IPCs) (Kanemura et al., 2014).
Nonetheless, iPSCs have many drawbacks and limitations. The main hurdles in the clinical application of iPSCs are the danger of teratoma formation, the mutagenic potential of introducing TFs, and the possibility of tumorigenesis (Yamanaka, 2020). Currently, the need to find new strategies based on reprogrammed cells to improve the safety of replacement therapies continues to be a key concern. The direct conversion of one somatic cell into another without the passage through pluripotent states is transdifferentiation. The direct reprogramming of a patient's somatic cells into pancreatic beta cells bypasses the induction process of stem cells, therapy to remove immune rejection, and the development of neoplasia (Ladewig et al., 2013). The mechanism of direct reprogramming is not completely known, but it has been recognized that epigenetic modulations by the ectopic expression of transcription factors or singling pathway modulators have important functions in the cycle of direct cell conversion (Zhu et al., 2014).
Because of the many programming weaknesses and disadvantages, including that some TFs and their implementation methods cause cancer, many previous studies have been performed using small molecules to enhance programming. Small molecules may encourage the efficiency of direct reprogramming based on TFs, and they often replace the effects of TFs, which is helpful in the definite and cost-effective preparation of a large number of cells. They are efficient and safe because they can be easily introduced and manipulated without genomic changes (Han et al., 2016).
The efficiency of direct reprogramming without the assistance of small molecules is fairly low. The use of small molecules in supporting this method has recently gained popularity. Small molecule-friendly direct reprogramming also results in far higher inefficiencies than those without the support of small molecules, and small molecules alone have also been used (Li et al., 2014). Therefore, this study aims to increase productivity and enhance the programming process by using small molecules to obtain insulin-generating cells from somatic cells (fibroblasts) and by using growth factors as an alternative to TFs.
Materials and Methods
This study was conducted at the Iraqi Cancer and Medical Genetic Research Center (ICCMGR).
Isolation, culturing, and propagation of skin fibroblast cells from newborn mice
Skin fibroblast culture was prepared as modified from Freshney (2015). Two- to 3-day-old new donor male Swiss albino mice were killed (ICCMGR/Animal House Unite). The newborn mice were killed, all limbs removed, skin cut, and minced into small pieces. Then these fragments were digested in 4 mg/mL collagenase type 1 (USBiological, USA) diluted in Minimum Essential Media (MEM) (USBiological) for 1.5 hours at room temperature on a magnetic stirrer. Then MEM was added with 10% fetal bovine serum (FBS), 1% penicillin, and streptomycin to stop enzymatic activity. After 10 minutes of centrifugation at 1500 rpm, the pellets were collected and washed twice in phosphate-buffered saline (PBS), resuspended in MEM 10%–20% FBS in a T-25 cm3 flask (SPL, Korea), and the cultures were maintained at 37°C in air humidified at 95% in a 5% CO2 incubator until cell confluence.
Subculturing was achieved after 3 to 4 days of cell culture and monolayer acquisition to avoid the contact inhibition of growth and spontaneous differentiation (Solchaga et al., 2004). The cells were discarded using 1–2 mL of 0.25% Trypsin/EDTA solution (USBiological, USA). Passage (1 and 2) was used in all experiments at 1 × 106 cells/mL.
Immunophenotypic analysis of skin fibroblast cells
Skin fibroblast cells (P1) were cultured in an eight-well tissue culture chamber slide (SPL) in MEM media with 10% FBS. Within 24 hours, the cells were allowed to grow a monolayer of adherent cells. The medium was then pipetted, and the multiwall plates were washed twice with PBS, fixed with cold acetone for 2–5 minutes, washed with PBS, and left to dry. These slides were used in triplicate. Four specific fibroblast cell markers were used: CD44, CD26, CD106, CD10, and two for stem cells, CD34 and vimentin (1:50) (Santa Cruz, USA). The slides containing samples were incubated for 10 minutes in a humidified chamber with 1% hydrogen peroxide (H2O2) after fixation, washed two to three times with a buffer wash, and incubated at room temperature with 2%–3% serum block for 30–40 minutes.
In a humidified chamber, the CD markers' primary antibody was incubated overnight at 4°C, but the control group was incubated with blocking serum without primary antibodies. They were then stained using an ABC Immunostain Mouse kit (Path Situ, India). The polyExcel target binder was incubated and washed for 10 minutes. PolyExcelpolyHR was applied and then incubated and washed for another 10 minutes. Finally, it was stained for 5 minutes with DAB prepared from 1 mL 3,3′-diaminobenzidine (DAB) substrate buffer and one drop of DAB stun substrate chromogen, thoroughly washed for 30–60 seconds with a washing buffer, and stained with hematoxylin. The control cells were stained without the primary antibiotic. The slides were mounted on a dibutylphthalate polystyrene xylene (DPX), analyzed, and captured using a light microscope and a digital camera (Moral-Sanz et al., 2012).
Transdifferentiation of skin fibroblast cells into IPCs in vitro
Using certain growth factors and small molecules, trypsinization cells were cultivated in a 25 cm2 tissue culture flask at a density of 1 × 106 cells. To induce IPC differentiation by using four different differentiation induction media, the confluent cultures of P1 and P2 skin fibroblast cells were used as follows:
Induction media 1 (M1)
This method consists of four step formulas according to Majeed et al. (2015). Step one formula: the cell monolayer was treated for 24 hours with high-glucose MEM (H-MEM, 25 mol/L glucose) supplement containing 10% FBS and 10-6 mol/L retinoic acid (RA) (Santa Cruz Biotechnology, Inc.). Step 2 formula: after 10% FBS for 48 hours, the medium was changed to H-MEM. Step 3 formula: 10 mmol/L nicotinamide (Santa Cruz Biotechnology, Inc.), 20 ng/mL epidermal growth factor (EGF), and 20 ng/mL fibroblast growth factor (FGF) (USBiological USA).
The 6-day low-MEM medium with 10% FBS was changed. Step 4 formula: the medium was changed to a low-MEM supplement with 10% FBS and 10 nmol/L exendin-4 for 6 days. The other methods were based on the first approach, but they were modified by adding other molecules. The first difference in time and duration of addition was used to determine the best method and depend on it.
Method 2 (M2)
This also has four steps; the first step formula, which was updated by Majeed et al. (2015), consisted of an H-MEM containing 10% FBS and 10-6 mol/L RA, 100 ng/mL activin A (AA), 3 μmol/L (PEPRO TECH, USA), CHIR 99021 (USBiological, USA), and 5 μmol/L BRD 7552 (USBiological). The cells were incubated for 24 hours in this formula. The remaining steps (2–4) were identical to those indicated in method (M1).
Method 3 (M3)
This method consisted of two steps, each of which consisted of the following formulas. Formula 1: the cell monolayer was treated for 6 days with an H-MEM (25 mol/L glucose) supplement containing 10% FBS and 10-6 mol/L RA, 100 ng/mL AA), 3 μmol/L CHIR 99021, and 5 μmol/L BRD 7552. Formula 2: the medium was changed to low-MEM supplement with 10% FBS and 5 μmol/L BRD 7552, 10 mmol/L nicotinamide, 20 ng/mL EGF, 20 ng/mL FGF, and 10 nmol/L exendin-4 for 6 days.
Method 4 (M4)
It consists of five steps. Formula of step 1: this formula consisted of 10% FBS MEM media and 1 μmol/L Bix (Santa Cruz Biotechnology, Inc.). Step 2 formula: the medium was changed to a 10% FBS MEM supplement with 100 ng/mL AA and 5 μmol/L CHIR 99021 for 72 hours. Step 3 formula: the old medium was extracted and incubated in new MEM media with 10% FBS, 10-6 mol/L RA, 5 μmol/L BRD 7552, and 20 ng/mL FGF for 4 days. Step 4 formula: the previous medium was changed to a new MEM supplement with 10% FBS and 20 ng/mL EGF for 5 days. Step 5 formula: the medium was modified to 10% FBS MEM supplement with 10 mmol/L nicotinamide, 10 nmol/L exendin-4, and 20 ng/mL FGF for 7 days.
Morphological study
Hematoxylin and eosin (H&E) staining has been used in many morphological studies. Undifferentiated skin fibroblasts and induced or differentiated cells were cultured at 1 × 106 cell per plate on the cover slide in a six-well tissue culture plate. After 3 days, the medium was pipetted, and 10% formaldehyde (PBS diluted) was added for 30 minutes to fix the cells. The cells were washed and dehydrated in absolute ethanol at various concentrations (100%, 90%, and 70%), each treatment lasting for 2 minutes.
Next, the cells were washed twice in distilled water (DW). The cover slide was then stained with hematoxylin (as a nuclear stain) for 2 minutes. Then the cells were washed twice in DW. Subsequently, the slide was stained with eosin for 2–5 minutes (a cytoplasmic stain), and the cells were washed twice with DW. The cells were then dehydrated for 5 minutes with absolute 100% ethanol (Safi et al., 2019). Finally, the cells were mounted using DPX and photographed with a camera using a light microscope (Micros, Austria).
Insulin release assay by ELISA
Glucose-stimulated differentiated cell insulin release (in four induction media) was incubated with serum-free media (SFM) containing 27.7 mM glucose at 37°C for 90 minutes and then changed to SFM containing 2.5 mM glucose for 15 minutes at 37°C. With 5.5 mM glucose and only SFM, as adapted from Shi et al. (2005) and Anjum et al. (2018), the control (undifferentiated cells) was incubated for 15 minutes. Then the related conditioned supernatant was collected and analyzed. The insulin concentration was determined using a mouse insulin ELISA kit (Elabscience, USA) as recommended by the manufacturer.
Dithizone staining
The differentiated clusters of IPCs were examined by staining with dithizone (DTZ) from day 16 of culture. The stock solution was prepared as in Mehrabi et al. (2015) by dissolving 50 mg of streptozotocin (STZ) (Santa Cruz Biotechnology, USA) in 50 mL of dimethyl sulfoxide (Santa Cruz Biotechnology) and stored at −20°C. By applying10 μL of dye stock solution to 1 mL of culture media, in vitro DTZ staining was achieved. Both differentiated and undifferentiated cells were incubated in a medium containing DTZ at 37°C for 30 minutes.
Detection of pancreatic protein expression through the transdifferentiation process
The cells were fixed at different periods (1, 3, 9, and 16 days) using the induction method for the immunocytochemistry (ICC) assay for each Oct3/4, stemness marker, insulin, c-peptide, and glucagon (Santa Cruz Biotechnology, Inc.). The cells were fixed and stained using the Immune Staining System Kit (Path Situ, India) at all times. Staining was done using specific antibodies with Oct3/4 as a stemness marker and insulin, c-peptide, and glucagon as mature pancreatic markers. The ICC assay was conducted according to the manufacturer's instructions.
Quantitative image analysis
Digital images of ICC were analyzed through the quantitative analysis protocol for counting the stained hematoxylin-DAB staining slides. Images were taken by light microscope and camera (Micros System, Austria). In this study, three separate staining areas of ICC images of each slide were analyzed: (1) the deconvolution color technique was used to un-mix the DAB, and the hematoxylin-stained areas left a complimentary image (Schindelin et al., 2012); (2) three DAB images were selected, and the intensity of cells was calculated using the Fiji version of ImageJ (http://Fiji.Sc) (Alawsi et al., 2019); (3) in the results window, the intensity number was translated to an optical density (OD) using OD = log (max intensity/mean intensity), max = 255 in 8-bit images (Mustafa et al., 2015).
IPC supernatant injection into diabetic mice
The mice were kept in standard conditions. All the experimental procedures were approved by the scientific committee of the Iraqi Center for Cancer and Medical Genetics Research, Mustansiriyah, Baghdad, Iraq. Experimental diabetes was induced by a single i.p. STZ injection (Santa Cruse, USA), 200 mg/kg freshly dissolved (1.47 g sodium citrate in 50 mL deionized water) at pH 4.66 (Islam and Code, 2017) in male Swiss albino mice aged 8–10 weeks. The mean blood glucose level was 419.91 ± 69.68 mg/dL 6–7 days after the injection of STZ.
Five animals were injected with 500 μL of M2 IPC culture supernatant; three animals were injected with 500 μL of SFM; three animals were injected with 500 μL of noninduction media; and three animals were injected with 0.25–1.5 U/mL of insulin (Schaschkow et al., 2016). The negative controls were not injected. Blood glucose levels were determined before and after injection at 30, 60, and 120 minutes using an Accu-Chek glucometer (Germany).
Statistical analysis
The data were analyzed using GraphPad Prism version 6.07 for Windows (GraphPad Software, San Diego, CA). They were shown as mean ± SD (n = 3). One-way analysis of variance (ANOVA), two-way ANOVA, and multiple comparisons were used to obtain all results (significance at p < 0.05 and p < 0.01).
Results
Culturing, immunophenotypic characterization of mice skin fibroblasts, and digital image scoring
The skin fibroblasts collected from the Swiss albino mice were cultured in tissue culture flasks for 18–20 hours; a few adherent cells were attached, and some colonies were observed. The first medium shift was discarded with no adherent cell and debris, usually at 24 or 48 hours (Fig. 1A). It was observed that adherent cells started to proliferate 2–3 days after the cultivation to form multiple fibroblast-like cells. Skin fibroblasts are distinguished by their ability to form colonies of spindle-shaped cells derived from a single cell. The cells expanded to more than 80% confluence at 3–4 days and were ready to be passaged. In passage one (P1), the skin fibroblasts started to expand, and a homogeneous layer of spindle-like shaped cells filled the entire plastic surface (Fig. 1B). These cells represented undifferentiated cells (Time 0) before their differentiation.

Characterization of mice skin fibroblast.
In addition, the morphological features of the cultured fibroblasts showed spindle-shaped cells and an oval-shaped nucleus.
Cultured skin fibroblasts were fixed and tested against a panel of stem cells and fibroblast-related surface antigens: CD34, CD106, CD10, CD26, CD44, and vimentin. The immunophenotypic characterization of expanded adherent cells showed that the cells were stained negatively for CD34 and CD106 (Fig. 1C, D), implying that these cells were not of hematopoietic and MSC origin. However, they were positive for each of CD10, CD26, CD44, and vimentin (Fig. 1E–H). These results indicated that the cells maintained the skin fibroblast phenotype. Whether the CDs were negative or positive, all the CDs characterized by fibroblasts showed a significant difference compared to the control (Fig. 1I).
Transdifferentiation of skin fibroblasts into IPCs and evaluation of the best insulin-producing induction media
The first passage (P1) of skin fibroblasts was used to induce IPCs using four different Formulas (induction media). Formula 1 (at day 1 of exposure) was used to culture skin fibroblasts in H-MEM supplemented with 10% FBS, AA, CHIR 99021, RA, and BRD. AA and CHIR 99021 are small molecules that induce the differentiation of stem cells into definitive endoderm and initiate epigenetics activation (Li et al., 2014). Formula 2 was used to culture skin fibroblasts cells in H-MEM supplemented for 2 days with 10% FBS. In Formulas 1 and 2, BRD and RA with high-glucose media also induced cells to produce pancreatic precursors, in addition to cells to the definitive endoderm by using AA and CHIR 99021 (Cai et al., 2010).
In Formula 3, the skin fibroblasts cells were cultured for 6 days in a low-glucose MEM supplement of 10% FBS with nicotinamide, FGF, and EGF, which induced pancreatic cells. Formula 4 was used to culture the cells in low-glucose MEM 10% FBS with exendin-4 to induce and form cells similar to IPCs.
Increasing quantities of insulin were discharged by clusters of differentiated cells at glucose levels of 27.7 Mm, as shown in Figure 2. The mean insulin concentration secreted by the clusters was 223.7 ± 2.49 pg/mL in M2.

Comparison analysis for insulin quantity secreted by induced cells (M1), (M2), (M3), (M4), (no induction), and (SFM) using ELISA assay. ****Indicates high significance p < 0.0001, and ns indicates no significance. SFM, serum-free media.
However, the mean was 8.429 ± 1.23 pg/mL in M1, 9.666 ± 1.23 pg/mL in M3, and 17.25 ± 1.23 pg/mL in M4. The cultured skin fibroblast supernatants (no induction) and SFM showed exceptionally low, barely detectable amounts of insulin, which could be disregarded (1.38 ± 2.46 and 0.783 ± 1.23 pg/mL, respectively). These results represented the experimental mean: the statistically significant difference between M2 and M1, M3, M4, no induction media, and SFM was statistically high (adjusted p-value <0.0001). Moreover, there was no significant difference (ns) between other induction media, except M1. The results showed that M2 was the best, so it was chosen and studied closely by analyzing morphological changes, detecting pancreatic protein expression, and characterizing the function of induced cells in vivo.
Morphological changes through transdifferentiation of skin fibroblasts into IPCs using M2 formula
The second induction media, M2, was the best inducer for insulin; therefore, we studied all the morphological changes during all the differentiation steps. Skin fibroblast cells exposed to step one formula showed no significant cell shape changes (Fig. 3A, B). In step 2, which lasted for 2 days, the cells start to enlarge after 3 days (Fig. 3C, D). During step 3, the cells were cultured for 6 after 9 days, which induced pancreatic cells. In this step, the cells started to aggregate in clusters. There were several cells that were defined by their elongated shape, as shown in Figure 3E and F. Finally, for another 6 days after 16 days, step 4 induces and forms cells similar to IPCs, which were represented by their small polygonal shape and the presence of clear and large clusters (Fig. 3G, H).
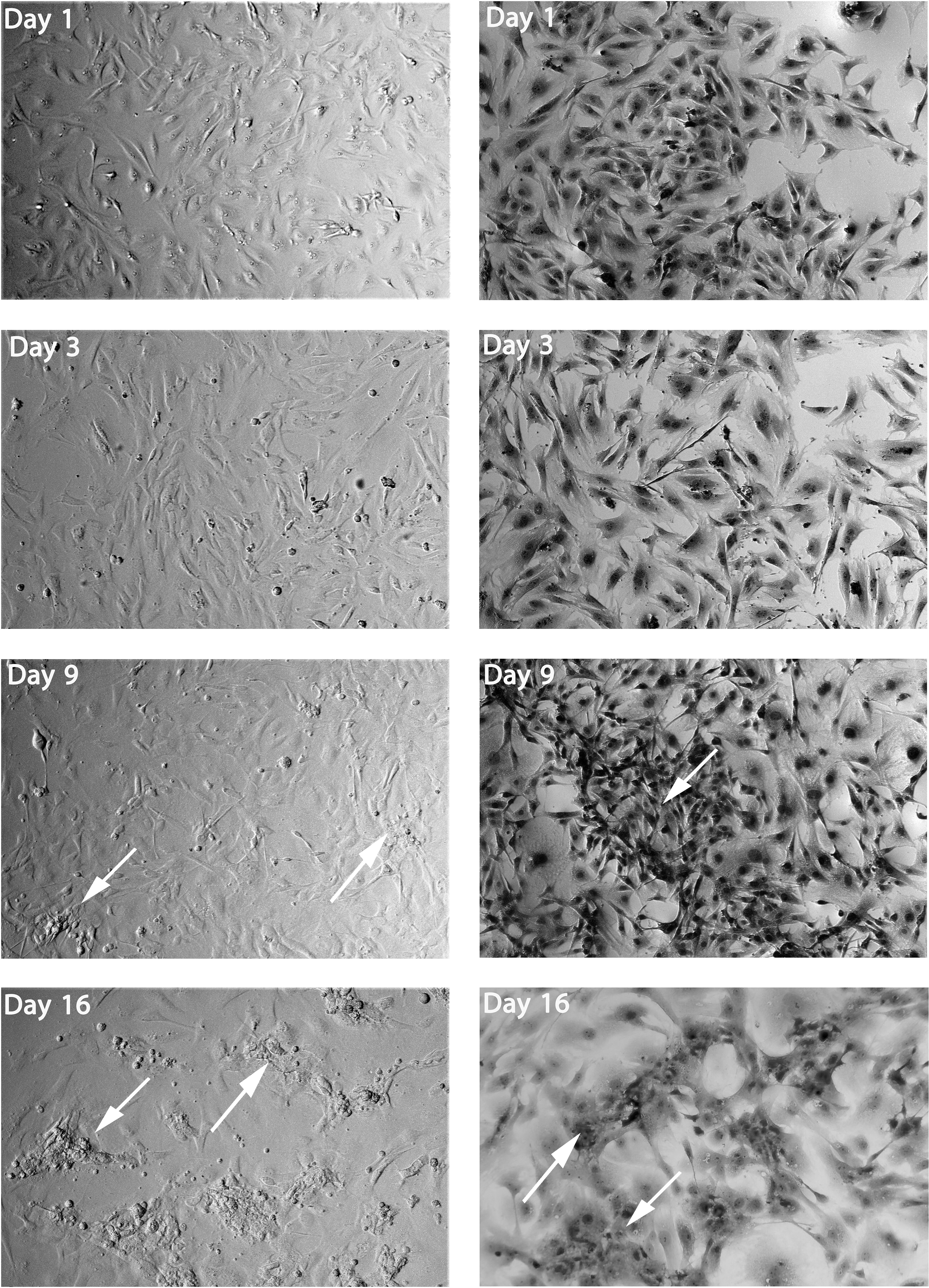
Skin fibroblasts after induced differentiation by induction media 2 (M2), showing the elongated shape of skin fibroblasts toward forming the IPC clusters. At day 1, cells showed no significant morphological changes. After 3 days, the cells start to enlarge. After 9 days, cells replaced by a polygonal shape with few clusters start to form (dark arrows). After 16 days, the IPC cluster formation to dominate the fields (white arrows). First column, no staining, second column stained with H&E, 20 × . IPC, insulin-producing cells.
Detection of pancreatic protein expression at different period of transdifferentiation process:
The results of the ICC analysis showed increased protein expression levels in each insulin, c-peptide, glucagon, and Oct3/4 marker as the time exposure of induction media 2 (M2) increased (1, 3, 9, and 16 days) as shown in Figure 4A, with differences in significance at p < 0.05 and p < 0.01. The cells in step one were exposed to AA, CHIR 99021, RA, and BRD for 1 day to promote endoderm commitment and induce pancreatic differentiation. Insulin, c-peptide, and glucagon were not yet expressed at this stage, but Oct3/4 markers began to express compared with the undifferentiating skin fibroblasts in the negative control. Figure 4 shows insulin, c-peptide, glucagon, and Oct3/4. Insulin, c-peptide, and glucagon started to express during step 2 formula exposure, but Oct3/4 expression increased compared with the first day of exposure and the negative control (Fig. 4C) shows insulin, c-peptide, glucagon, and Oct3/4.

Detection of pancreatic proteins in skin fibroblasts through transdifferentiation process induced by induction media 2 (M2), the ICC assay showed gradual increase starting on day 3.
The expression of pancreatic proteins (insulin, c-peptide, and glucagon) increased significantly, and the third step formula, consisting of low-glucose MEM 10% FBS/nicotinamide, FGF, and EGF exposure, was further stimulated and contributed to the development of clusters also; in this step, the expression of Oct3/4 increased significantly compared to the expression of oct3/4 at 1 day and 3 days and with negative control, indicating the stage of stemness as seen in Figure 4C. Finally, on day 16, the cluster development increased significantly and was triggered by the fourth step formula, which consisted of low MEM supplemented with 10% FBS/exendin-4. Compared with the expression of insulin, c-peptide, and glucagon at 1, 3, and 9 days and negative control, the expression of these markers increased, whereas, when compared to 9 days, the expression of oct3/4 decreased on day 16 of exposure (Fig. 4C).
IPCs identified by DTZ
The transdifferentiated cells were stained with the zinc-chelating agent (DTZ), which is a binding material, to test IPCs in the cultures. At the end of the experiment, the differentiated cell clusters were colored red crimson by the DTZ (Fig. 5A, B).
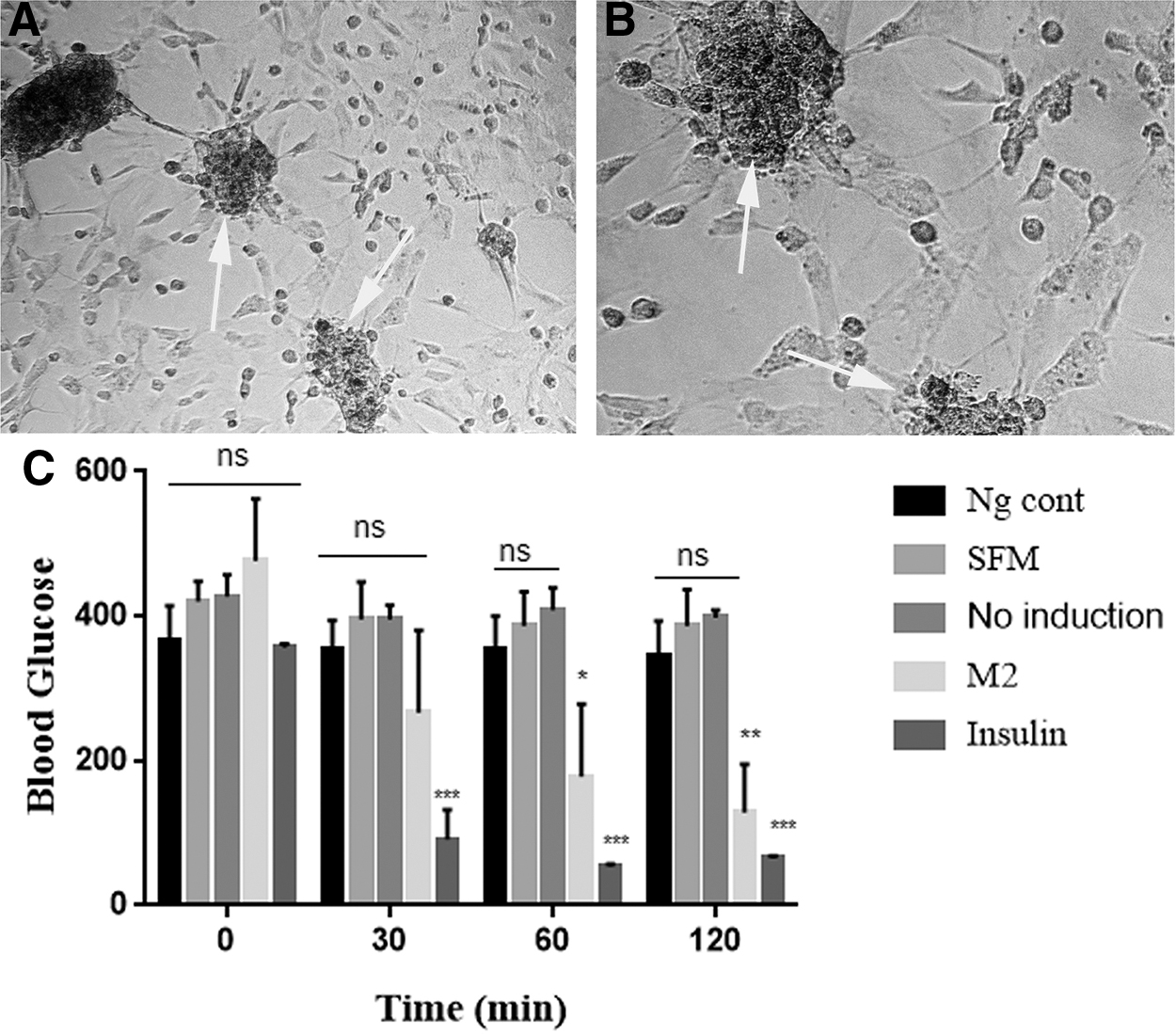
IPC clusters at 16 days of exposure shown to produce functional insulin that is able to lower glucose levels.
IPC supernatant was able to reduce glucose levels in diabetic mice
We were successful in obtaining IPCs in STZ diabetic mice by proving their functionality in supernatant cells that had been differentiated by M2.
To further evaluate the functional efficacy of insulin produced by transdifferentiated cells, 500 μL (223.7 pg/mL of insulin concentration) of supernatant for differentiated cells (M2) was injected by i.p. into diabetic mice at 16 days of exposure. Undifferentiated cell supernatant (no induction) and SFM were also injected to diabetic mice as controls. The results showed that the supernatant (M2) acted similar to insulin, reducing the blood glucose level in diabetic mice at 30, 60, and 120 minutes significantly. For other groups, glucose levels remained almost constant at different times at p < 0.05 and p < 0.01 (Fig. 5C).
Discussion
Transdifferentiation is the biological process that converts mature cells into other mature cells without passing through the pluripotent stage. It is used in cell therapy to reduce or prevent teratoma, tumorigenesis, and ethical religion through differentiated stem cells and induce pluripotent stem cells, which provide a promising treatment for various difficult diseases such as diabetes.
In several previous studies, costly enzymes have been used to separate the fibroblasts found in the dermis from the epidermis by applying more than one isolation technique (Choi et al., 2017; Li et al., 2017; Seluanov et al., 2010). In our experiment, we were able to isolate and culture fibroblasts using a quick and simple method. The results of a morphological study confirmed fibroblast nature, and ICC showed that the isolated cells had positive CD markers: CD44, which is a special marker of fibroblasts, and MSCs (Kundrotas, 2012; Nilforoushzadeh et al., 2017). CD10 and CD26 are highly positive in fibroblasts, but bone marrow mesenchymal stem cells (BM-MSCs) show less than 35%–40% expression (Cappellesso-Fleury et al., 2010). Vimentin has often been referred to as an intermediate fibroblast filament, and it is a rare and sufficient fibroblast marker that is distinguished by its mesodermal origin (Cappellesso-Fleury et al., 2010; Pereyra-Bonnet et al., 2014).
Other studies have shown that dermis cells called skin-derived precursor cells (SKPs) in skin-derived precursors were positive, expressed high fibronectin and nestin levels, and were negative for mesodermal markers such as vimentin (Pereyra-Bonnet et al., 2014). However, CD106 and CD34 produced negative results. In CD106, BM-MSCs were strongly positive, whereas fibroblasts were negative (Halfon et al., 2011; Nilforoushzadeh et al., 2017). The isolated cells in the skin's dermis were not of hematopoietic origin and MSCs because CD34 is a special and positive marker of hematopoietic stem cells, and it is positive in mouse MSCs located in the dermal skin bulge hair follicle, which Tempus first described in 2003 (Crigler et al., 2007; Morris et al., 2003).
This study demonstrated a simple and highly effective method for inducing fibroblast skin in mice for differentiation into in vitro IPCs. In the final stage, in vitro and in vivo assays showed that the features of the IPCs were similar to mature islet beta cells. We succeeded in directing skin fibroblasts efficiently into IPCs.
First, differentiated cells resulted from small molecules that served as an alternative or induce certain transcription factors that induce insulin output by mature cells. We used various induction media, the first of which was the best in differentiating skin fibroblasts into IPCs by using M2, as shown in Figures 2–4.
Second, at the final stage, supernatants of differentiated cells were assessed to determine insulin concentrations and injected by STZ into diabetic mice. After injection, the blood glucose level of diabetic mice was lowered, and this supernatant acted like insulin (Fig. 5). Because the supernatant media of the differentiated cell understudy were comparable to the action of insulin, this test was not previously mentioned in studies and research; it was used to confirm the conversion and differentiation of cells into IPCs.
Many of the research mentioned, as well as others such as Pennarossa et al. (2013) and Pereyra-Bonnet et al. (2014), Majeed et al., and Zhu et al. (2016), respectively, focused on the differentiation and transdifferentiation of cells from various sources into IPCs or beta cells in vitro (Moral-Sanz et al., 2012). Then, to cure diabetes, inject these cells into diabetic animals and find the best ways to differentiate and raise the differentiation rate to improve the efficiency of using these cells in diabetes treatment.
In the differentiation process, a method has been relied on in previous studies, namely Majeed et al. (2015), in which MSCs were utilized and differentiated into IPCs, and from this approach, modifications were made, and many methods were employed to find the best way for differentiation and approved in the study. The reason for using this approach is the similarities in morphology, cell surface markers, differentiation capacity, and immunomodulatory properties between MSCs and fibroblasts, which have been mentioned and confirmed in many studies, including Denu et al. (2016) and Ichim et al. (2018). As a result, fibroblasts and MSCs have similar immunomodulation processes, making fibroblasts an alternate source for regenerative therapy (Ugurlu and Karaoz, 2020). And it is also through our results that showed the similarity between fibroblasts and MSCs through some markers.
The M2 technique was used to generate IPCs from fibroblasts using four different formulas containing various chemicals and growth factors in this study. We used H-MEM supplemented with 10% FBS and the other chemicals specified above in formulas 1 and 2.
Glucose is a growth factor in beta cell replication at concentrations of 20–30 mol/mL. High glucose levels are essential for the endocrine pancreatic differentiation process in mammalian cells, and they seem to be essential for functional efficacy and morphological organization (Zenobi et al., 2016). Kim et al. (2016), Pennarossa et al. (2013), and Brevini et al. (2016) studied the role and importance of high glucose in the differentiation of pancreatic cells, showing that mammalian cells were not able to differentiate into insulin-secreting cells (ISCs) in a low-glucose environment.
However, they were able to differentiate ISCs in high glucose. AA is essential for early final endoderm production, and a combination of AA and CHIR 99021 improved and enhanced efficient activating genes and TFs more than their singular use or combined use with other endoderm development molecules (Brevini et al., 2016; Yabe et al., 2017).
Another crucial molecule in formula 1 to further facilitate pancreatic lineage differentiation, we used RA, which is an important signaling molecule in early embryonic pancreas development in both endocrine and exocrine cells by inducing the expression of PDX-1. It is one of the most important transcription factors that regulate the expression of the insulin gene (Spaeth et al., 2017). RA signaling is required for the development of the dorsal pancreas from the foregut endoderm. Treatment of cultured mouse and human ESCs may trigger the production of Ngn3 in endocrine progenitor cells and enhance beta cell differentiation (Lorberbaum et al., 2020).
Several studies clarified the importance of RA in differentiating definitive endoderm into pancreatic progenitor cells combined with other molecules or factors, including FGFs, EGFs, BRD, AA, Noggin, and others (Meng et al., 2018). Following the sequential gene expression of sufficient transcription factors, such as Ngn3, NeuroD, Nkx2.2, Pax4, Pax6, and MafA, Pdx1-expressing pancreatic precursor cells were induced to differentiate into IPCs (Kubo et al., 2011). BRD7552, another small molecule that induces and replaces PDX1, enhanced PDX1 expression in primary human islets and ductal cells and triggered epigenetic alterations in the PDX1 promoter consistent with transcription activation (Yuan et al., 2013). Zhu et al. (2016) showed that the transdifferentiation of fibroblasts into pancreatic-like cells involved a merged effect that increased the expression of pancreatic-specific genes and reduced the expression of fibroblast-specific genes by influencing epigenetics (the process of turning genes on and off).
Kubo et al. (2011) showed that the transdifferentiation of fibroblasts into pancreatic-like cells involved a merged effect that increased the expression of pancreatic-specific genes and reduced the expression of fibroblast-specific genes; however, there was no significant shift in transcriptome-wide scaling. Specifically, PDX1 participated in pancreatic endoderm induction, while NGN3, during pancreatic differentiation, induced the differentiation of pancreatic endocrine cells from epithelial progenitors (Zhu et al., 2017). This study's findings support the contribution of the co-expression of these two variables to a higher increase in the insulin gene than when the upregulation of only PDX1 occurs.
In the next step of our experiment, the expression of pancreatic proteins in formula three increased compared with the previous step (Fig. 3), which was induced by the use of various compounds, including nicotinamide, EGF, and the FGF. EGF increased the PDX1 expression of mouse ESCs (Lorberbaum et al., 2020; Majeed et al., 2015). FGF enhanced cell proliferation and the early stages of pancreatic precursor cell clustering (Aigha and Abdelalim, 2020). Nicotinamide promotes reprogramming, improves cell maintenance, and facilitates cell differentiation into many lineages, including pancreatic and other lineages (Griffin et al., 2017). Nicotinamide has the ability to increase MafA expression, which is an important factor in beta-cell formation and maturation, and increasing MafA expression in IPCs may improve cell maturity (Aigha and Abdelalim, 2020).
In the last step of our experiment, the results showed the increased expression of pancreatic proteins, especially insulin, which was induced by exendin-4 that increases the biosynthesis of glucose-stimulated proinsulin and stimulates the process of insulin-mediated glucose uptake (Arakawa et al., 2009). Exendin-4 enhances the differentiation of IPCs through activation of various β cell markers such as Pdx-1, Nkx2.2, and MafA (Kassem et al., 2016).
Finally, as shown previously, many hormone-positive in vitro cells obtained in this study co-expressed glucagon, insulin, and somatostatin, suggesting a polyhormonal immature state in vitro (D'Amour et al., 2006; Pennarossa et al., 2013; Zhu et al., 2016). In our study, as shown in Figures 3 and 4, in the process of converting and differentiating fibroblasts into IPCs using induction media in different periods, it was observed that the cells passed through a stemness stage, especially at 9 days, when the cell shape and protein expression of the OCT3/44 marker showed increased peaks. Many previous studies described the development of pancreatic beta cells from fibroblasts and showed evidence that the reprogrammed cells did not move through a pluripotent stage (Pennarossa et al., 2013; Pereyra-Bonnet et al., 2014).
However, Bar-Nur et al. (2015) and Maza et al. (2015) found that the majority of somatic cells had to pass through a transient pluripotent state marked by oct4 expression when the somatic cells were reprogrammed using iPSC reprogramming factors, which indicated that it was not entirely clear whether the fibroblasts passed through a short pluripotent stage before β cell reprogramming.
Conclusion
Using small chemical molecules and growth factors in vitro, we developed a simple and fast method for transdifferentiating mice skin fibroblasts into IPCs. The induced cells passed through a transient pluripotent stage during the transdifferentiation process before forming IPCs. The differentiated cells secreted measurable quantities of glucose-dependent insulin that were assessed for functionality in vivo in diabetic mice. The efficient conversion of readily available fibroblasts into IPCs without the need for gene transfection could lead to a promising therapy for diabetes and the generation of cells, which was difficult to achieve.
Footnotes
Authors' Contributions
Conceptualization of this study was done by A.M.A-S. Methodology was developed by A.M.A.-S and I.S.S., validation was done by A.M.A.-S., and M.K.H., A.M.A.-S., and I.S.S. conducted formal analysis, investigation, data curation and visualization, and drafting of the article. M.K.H. and A.M.A.-S. provided resource, supervision, and project administration. All authors revised and approved the article.
Acknowledgments
The authors would like to thank the staff of the Experimental Therapy department, Iraqi Center for Cancer and Medical Genetic Research, Mustansiriyah University, for their support during the work.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.