
Abstract
Tumor tissue comprises a highly complex network of diverse cell types. The tumor microenvironment (TME) can be mainly subdivided into cancer cells and stromal cell compartments, the latter include different types of immune cells, fibroblasts, endothelial cells, and pericytes. Tumor cells reprogram immune cells and other stromal cells in the TME to constrain their antitumor capacity by creating an immunosuppressive milieu and metabolism competition. Moreover, the reprogramming effect on immune cells is localized not only in the tumor but also at the systemic level. With wide application of single-cell sequencing technology, tumor-specific characteristics of immune cells and other stromal cells in the TME have been dissected. In this review, we mainly focus on how tumor cells reprogram immune cells both within the TME and peripheral blood. This information can further help us to improve the efficiency of current immunotherapy as well as bring up new ideas to combat cancer.
Cellular and Noncellular Components in the Tumor Microenvironment
The tumor microenvironment (TME) is a heterogeneous system comprising a variety of cells. The composition of the TME differs between tumors. This microenvironment consists of proliferating tumor cells, endothelial cells, stromal cells, and fibroblasts that assist in tumor progression and metastasis (Giraldo et al, 2019). Tumor progression is largely determined by interactions between immune cells, nonimmune cells, and proteins in the extracellular matrix (ECM) (Giraldo et al, 2019). These processes include vascularization, alteration of the ECM, increased proliferation of fibroblasts, and reprogramming of recruited immune cells (Bhowmick et al, 2004).
Tumors also interact with proteins in the ECM, such as collagen, fibronectin, hyaluronan, and laminin, which determine metastatic ability (Baghban et al, 2020). Blood vessels create a dynamic structure that controls the flow of nutrients and various immune cells into tissues. Tumors induce angiogenesis by secreting vascular endothelial growth factor (VEGF) to create their supply of blood within the TME (Nagy et al, 2009). The density of blood vessels in the central region of the tumor tends to be lower compared with the outer regions (Kamoun et al, 2010).
Subsequently, as tumor size increases, blood vessel density in the tumor center decreases, creating hypoxic central zones (Nagy et al, 2009). Tumors undergoing hypoxia lead to a dysfunctional endothelium and leaky blood vessels (Dudley, 2012). Leaky blood vessels involve the disruption of endothelial cell junctions, increasing the chances of tumor intravasation and metastasis (Peng et al, 2019). Fibroblasts favor angiogenesis, invasion, and cell proliferation in the TME through production of tumor-supporting ECM (Bhowmick et al, 2004).
In the TME, recruited stromal cells are reprogrammed into tumor-associated stromal cells (TASCs) (Bussard et al, 2016). TASCs show an increased expression of α-smooth muscle actin, fibroblast-activating protein, and matrix metalloproteinases. TASCs also secrete interleukin (IL)-6, IL-8, VEGF, and stromal-derived factor-1α, which can initiate the recruitment of tumor cells (Bussard et al, 2016).
Immune cells in the TME can be categorized either as immunosuppressive or immunostimulating cells (Palucka and Coussens, 2016). Immunosuppressive cells include tumor-associated macrophages (TAMs), regulatory T cells, regulatory B cells, immature dendritic cells (DCs), immature granulocytes, and monocytes. Immunostimulating cells include CD8+ and CD4+ T cells, natural killer cells, cytotoxic macrophages, and cytotoxic neutrophils (Salemme et al, 2021). The immunosuppressive cells allow tumor cells to avoid both innate and adaptive immune surveillance, resulting in proliferation and metastasis of cancer cells (Gonzalez et al, 2018).
There are different methods of further categorizing immune subtypes in tumors. One study performed a cluster analysis on 30 cancer types to identify six immune subtypes based on differences in macrophage and lymphocyte signatures, Th1:Th2 ratios, intratumoral heterogeneity, aneuploidy, and expression of immunomodulatory genes (Thorsson et al, 2018).
The six subtypes are (1) wound healing tumors that show a bias toward increased infiltration of Th2 cells involved in adaptive immunity; (2) the interferon (IFN)-γ dominant subtype that shows an increase in M1/M2 polarization and T cell receptor diversity with strong CD8+ signals; (3) the inflammatory subtype where an increase in the expression of Th17 and Th1 genes can be found; (4) the lymphocyte-depleted subtype that suppresses Th1 cell infiltration while expressing an increase in the presence of TAM of the M2 type; (5) immunologically quiet tumors that show the lowest lymphocyte levels and highest response of macrophages of the M2 type; and (6) the TGF-β dominant subtype with the highest levels of TGF-β expression and increased lymphocytic infiltration where the distribution between type 1 and type 2 T cells is equal (Fig. 1).
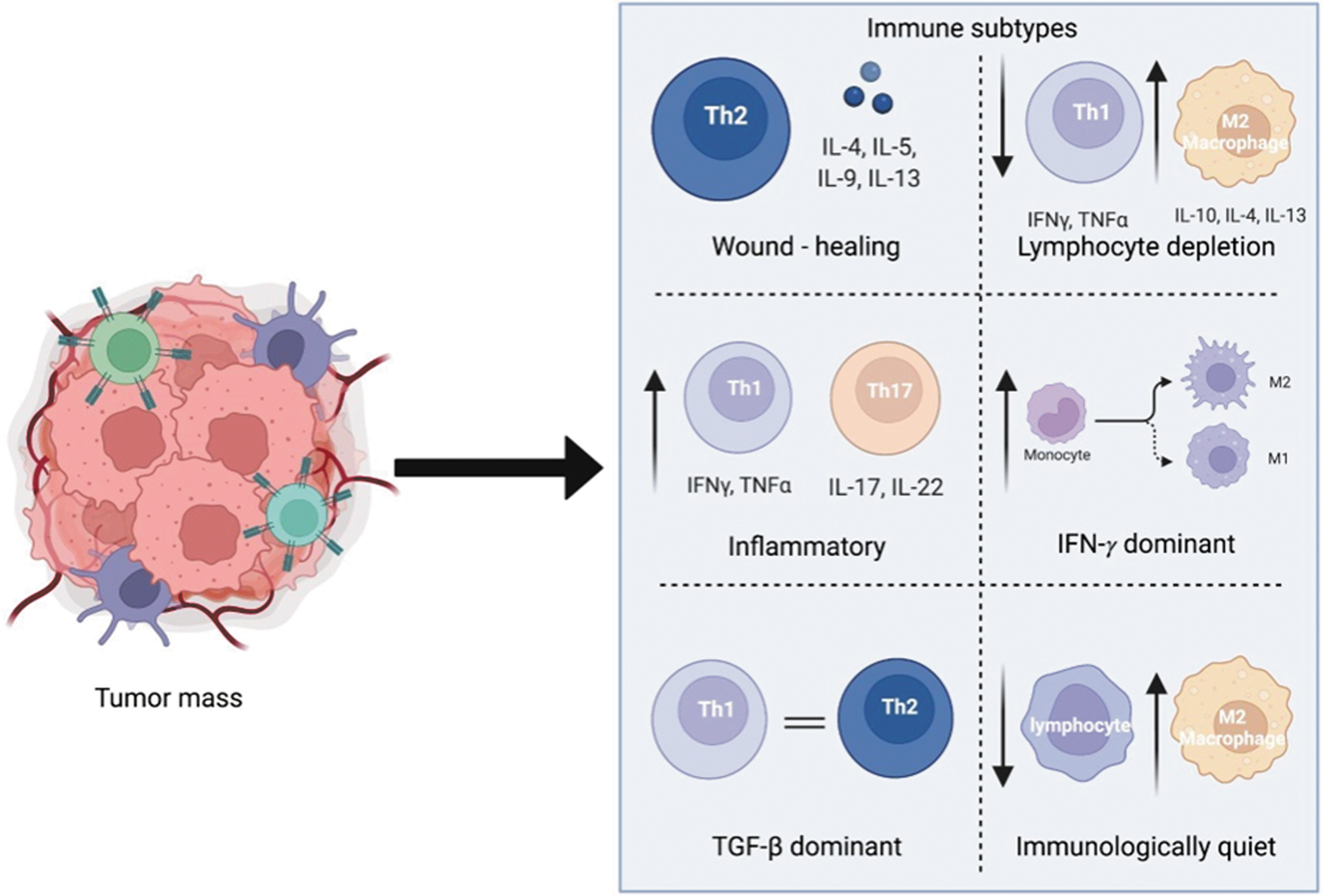
The six immune subtypes of the TME. (1) Wound healing subtype with increased Th2 bias. (2) Lymphocyte-depleted subtype, with depletion favoring an increase in anti-inflammatory macrophages and a decrease in Th1 cells. (3) Inflammatory subtype, expressing an increase in both Th1 and TH17 cells. (4) Interferon-γ dominant subtype, showing an increase in macrophage polarization in conjunction with increasing T cell receptor diversity. (5) TGF-β dominant subtype, where the ratios of type 1 and type 2 T cells are equal. (6) Immunologically quiet subtype, which consists of an increased presence of anti-inflammatory macrophages and decreased presence of lymphocytes. TGF, tumor growth factor; TME, tumor microenvironment.
Immune states can also be characterized by analyzing gene expression signatures of the same tumor type to identify heterogeneity. A study on intrahepatic cholangiocarcinoma resulted in the identification of four immune subtypes instead of the abovementioned six immune subtypes (Bagaev et al, 2021; Job et al, 2020). The four immune subtypes were identified to provide a more generalized view of the presence and role of immune cells in the TME.
The four subtypes are (1) immune desert where the gene expression signatures of tumor and stromal immune cells are very weak, suggesting downregulation of most immune pathways in the TME; (2) reactive immunogenic TME consisting of significantly high levels of innate/adaptive immune cells, quiescent hematopoietic stem cells, and activated fibroblasts; (3) myeloid-rich subtype expressing very high levels of monocyte-derived myeloid signatures and low levels of lymphoid gene signatures; and (4) mesenchymal subtype showing a significantly high level of expression for activated fibroblasts. The four immune subtypes were found in various tissue samples of the same type of cancer, suggesting some heterogeneity between patients.
Inflammatory and reactive immunogenic subtypes are predicted to have the most favorable outcome for immunotherapy based on the gene signatures of immune cells in the TME (Thorsson et al, 2018). An overall increase in inflammatory and innate immune cell infiltration into the tumor can support the pathways intended (by immunotherapy) for an increased survival rate and decreased tumor progression (Job et al, 2020).
As these subtypes are defined based on individual research methodology and parameters, researchers' understanding of tumors and their sensitivity to treatment also differs. However, a consensus can be reached that tumors that are more immunogenic and inflammatory would theoretically respond better to immunotherapy (Kim et al, 2020). It may also assist in deciding which type of immunotherapy would be best suited for the TME of that particular patient (Bagaev et al, 2021). These categories give us a general idea of the type of immune landscape that can affect how tumors are perceived and treated.
Immune Cell Reprogramming in the TME
The TME actively suppresses immune cells (Fig. 2) (Kurten et al, 2021). This section will address the types of immune cells and the mechanisms of their suppression.
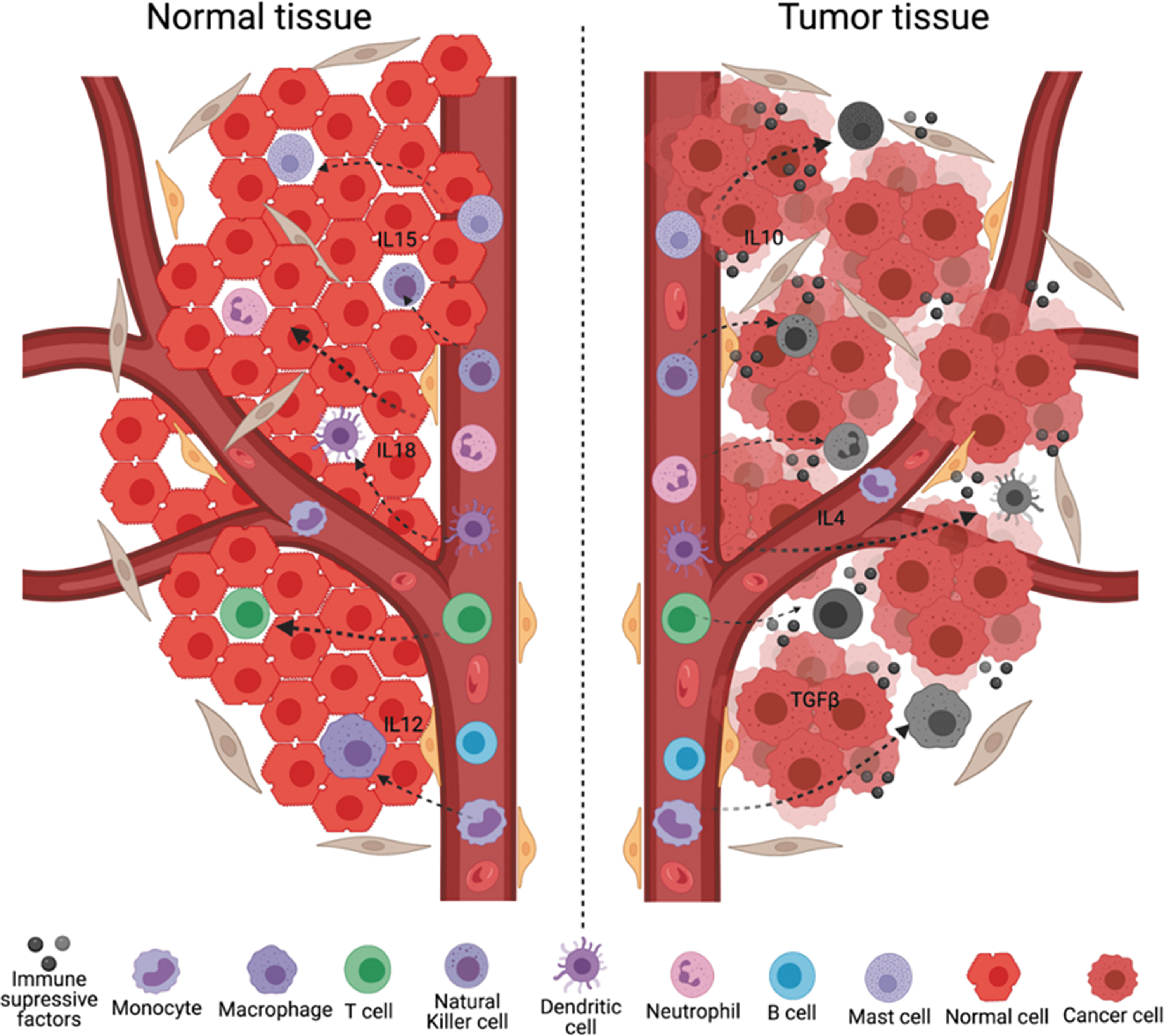
Immune cells are suppressed in the TME. Immune cells (T cells, monocytes, macrophages, B cells, natural killer cells, dendritic cells, mast cells, and neutrophils) are reprogrammed to the immunosuppressive phenotype after engaging with tumor cells or tumor-secreted factors.
CD8+ cytotoxic T cells, the major workhorse of tumor killing, succumb to immunosuppressive mechanisms of the TME (Wu et al, 2021). Apoptotic dysfunctional CD8+ T cells in the TME are associated with poor efficacy of immunotherapy. The immunosuppressive TME expresses immune checkpoint PD-1 and downregulates expression of IFN-γ, both of which actively suppress CD8+ T cells (Sanmamed et al, 2021).
Tregs are the major culprits in cytotoxic T cell exhaustion. Tregs, specified by Foxp3+ from the single-positive (CD4+) stage in the thymus, enter the TME and prevent CD28 costimulation to reduce production of IFN-γ and tumor necrosis factor (TNF)α in neighboring cytotoxic T cells (Zappasodi et al, 2021).
Myeloid-derived suppressor cells (MDSCs), another major component, suppress immune cells in the TME. When macrophages are exposed to immunosuppressive factors such as IL-4, IL-10, and TGF-β, they polarize into antiproinflammatory macrophages, the so-called TAMs. The origin of TAMs is either tissue-resident macrophages or monocyte-derived macrophages from the bone marrow. The former support initiation of tumors and the latter support proliferation of cancer cells (Klemm et al, 2020).
Tissue-resident macrophages recruit Tregs to protect tumor cells in the early stage of lung cancer. Then, tissue-resident macrophages will be replaced by monocyte-derived macrophages that enhance tumor progression (Casanova-Acebes et al, 2021).
There are two types of DCs: conventional dendritic cells (cDCs) derived from myeloid progenitor cells and plasmacytoid dendritic cells (pDCs) derived from lymphoid progenitor cells. Based on the function and transcriptional marker, cDCs have cDC1 and cDC2 subsets. cDC1s express Irf8 and Zbtb46, which mainly interact with CD8+ T cells. Whereas cDC2s express Irf4 and Zbtb46, which engage with CD4+ T cells (Anderson et al, 2021).
IL-4 blocks cDC1 antitumor immunity by exhausting them. Blockage of IL-4 restores IL-12 production from cDC1s and sustains the antitumor immunity (Maier et al, 2020). Tumor-secreted TGF-β reprograms pDCs to secrete IL-10, it can also induce immune tolerance by suppressing the expression of IFN-α (Labidi-Galy et al, 2011; Mitchell et al, 2018).
Natural killer (NK) cells are large, granular cytotoxic lymphocytes. However, NK cells can be reprogrammed into the protumor phenotype in the TME. Tumor cells can induce NK cells to express CD73 through immune checkpoint receptors such as PD-1 and LAG-3 (Neo et al, 2020). NK cells were exhausted or became dysfunctional after engagement with human breast tumor-secreted immunosuppressive factors such as TGF-β. They were also exhausted through downregulation of DAP10 and upregulation of PD-1 in the TME (Bi and Tian, 2017; Mamessier et al, 2011). IL-10 upregulated the inhibitory receptor, NKG2A, to cause dysfunction of NK cells, and blocking IL-10 can restore the function of NK cells (Sun et al, 2017).
B cells become either plasma cells or memory B cells when activated. Plasma cells secrete specific antibodies to target antigen-expressing tumor cells. However, tumor-infiltrating B cells in the TME secrete TGF-β and reprogram CD4+ T cells into FoxP3+ Treg cells. Hence, B cells enhance tumor metastasis (Guo and Cui, 2019; Olkhanud et al, 2011).
Neutrophils are the fastest responders toward the inflammatory site in the TME. Neutrophils release a degranulation protein and phagocytize the tumor with the help of other immune cells (Nielsen et al, 2021). Neutrophils are reprogrammed to immunosuppressive phenotypes that highly express LOX-1 in the TME (Nan et al, 2018; Shaul and Fridlender, 2019). Mast cells stimulate production of VEGFs to promote tumor growth through angiogenesis (Beer et al, 2008).
Transcriptome Diversity and Metabolic Competition Direct Immune Cell Reprogramming
Tumor cells reprogram immune cells and other stromal cells by forming a strong immunosuppressive tumor milieu to suppress the antitumor ability of immune cells. The TME significantly enriches CD8+-exhausted T cells with reduced cytotoxic activity and increased expression of inhibitory receptors compared with normal tissues. According to a single-cell RNA-sequencing analysis of gastric cancer, CD8+-exhausted T cells are transcriptionally diverse and differ from other CD8+ T cells in normal tissues (Sathe et al, 2020; Thommen and Schumacher, 2018).
However, CD4+ T cells and NK cells display transcriptional similarity between tumor and normal tissues, implying that the TME is particularly hostile to cytotoxic CD8+ T cells rather than other types of T cells and NK cells. A comparison between pancreatic cancer biopsies and matched organoids suggested that the TME controls the transcriptional diversity of cancer cells that define the response to chemotherapy. These observations indicate that the TME and immune cells in turn define the transcriptional signatures of each other, determining their response to therapeutics (Raghavan et al, 2021).
Liver cancers containing higher transcriptomic diversity were associated with patients' worse overall survival. Highly heterogeneous liver cancer cells produce Vascular Endothelial Growth Factor A (VEGFA) to vascularize the tumor and then recruit immune cells in the tumor. Consequently, tumor-infiltrating T cells exhaust and reduce cytolytic activities, indicated by reduced expression of granzyme and immune checkpoint molecules (Ma et al, 2019). SPP1 expression is tightly related to liver cancer cell evolution and microenvironmental reprogramming, indicating its use as a prognosis marker (Ma et al, 2021).
TAMs in gastric cancer tissue showed transcriptional diversity regulated by NFKB1, ETS2, CREM, REL, STAT1, and FOXO3 rather than a dichotomous M1/M2 view of macrophage activation states (Martinez et al, 2006). These TAMs showed different transcriptional characteristics from monocytes in peripheral blood, indicating an active reprogramming mechanism of macrophages in the TME. In human breast cancers, TAMs predominantly express apolipoprotein E, but not in monocytes and DCs.
A recent study identified that TAMs have two major categories. TREM2+ TAMs suppress immune cells. Meanwhile, the high number of FOLR2+ TAMs in the TME correlated with better patient prognosis (Nalio Ramos et al, 2022). The identification of immunostimulating FOLR2+ TAMs would lead to drug discovery that can reprogram TAMs to be immunostimulating. An immunosuppressive DC population with high expression of IDO1 and Treg predominates in the TME (Mellor and Munn, 2004; Sathe et al, 2020).
Plasma cells in normal mucosal tissues mainly express IgA, while they predominantly express IgG in the TME. B cells' antitumor function mainly relies on the IgG antibody isotype (Sharonov et al, 2020). These observations indicate that transcriptional diversity directs the reprogramming of immune cells and targeting the mechanisms holds a clue for new therapeutics.
To win the competition for limited nutrients between immune cells, tumor cells rewire their metabolic environment to meet the demands of increased nutritional expenditure, facilitating their survival, proliferation, and expansion (Wu et al, 2020). One of the hallmarks of cancer is the altered metabolic state of the TME that suppresses immune cells (Pavlova and Thompson, 2016). Understanding how the metabolic state suppresses immune cells is vital to advancing efficient cancer immunotherapy.
Tumor cells can siphon energy from T cells through nanotube-mediated transfer of mitochondria, resulting in suppression of T cell function (Saha et al, 2022). Tumor cells generate energy mainly through glycolysis and thus utilize glucose rapidly. Tumor cells outcompete immune cells in consumption of glucose, ergo inhibiting antitumor immunity (Chang et al, 2015; Sung and Cheong, 2022). Due to uncontrolled cell growth, cancer cells consume oxygen rapidly and create hypoxic conditions (Vito et al, 2020).
Hypoxia in the TME suppresses the TCR signal and CD28-mediated activation of T cells. On the other hand, hypoxia polarizes TAMs toward an anti-inflammatory phenotype, and hypoxic TAMs release factors that promote tumor growth, cancer immunosuppression, and angiogenesis (Henze and Mazzone, 2016; Murdoch and Lewis, 2005). Moreover, glycolysis by tumor cells accumulates lactic acid and creates an acidic immunosuppressive environment, which inhibits cytokine production by T cells and polarizes macrophages toward anti-inflammatory TAMs (Colegio et al, 2014).
Glutamine as an important metabolic fuel was predominantly consumed by cancer cells. Different tumor types show significant variability in their response to glutamine deprivation (Hensley et al, 2013). Cancers that prefer glutamine consumption, such as renal cancer, MYC-induced liver cancer, and cancer cells originated from intestinal mucosa, limit glutamine levels in the TME, which inhibits immune cell infiltration (Cluntun et al, 2017; Still and Yuneva, 2017; Zhou et al, 2017).
Glutamine metabolism affects the convening of MDSCs in the TME by regulating the LAP/G-CSF axis, suggesting that glutamine metabolism inhibitors could improve the efficacy of anti-PD1 and anti-CTLA4 (Zhu et al, 2022). Tumor cells and anti-inflammatory macrophages in the TME deplete arginine (Fletcher et al, 2015; Grzywa et al, 2020; Kostourou et al, 2011). A low concentration of arginine in the TME inhibits NK cell proliferation and IFN-γ production (Lamas et al, 2012).
The regulatory interaction between TME stromal and cancer cells is bidirectional. Cancer-associated fibroblasts (CAFs), as the major stromal cell type in the TME, significantly affect cancer cell metabolism. CAFs enact field cancerization by facilitating extratemporal oxidative stress (Liao et al, 2019).
Polarized macrophages display different modes of glucose metabolism. Proinflammatory macrophages acquire energy mainly by glycolysis and the pentose phosphate pathway (Dai et al, 2020; O'Neill and Pearce, 2016), while anti-inflammatory macrophages are sustained by the TCA cycle and mainly use oxidative phosphorylation (OXPHOS) (Sung and Cheong, 2022; Yu et al, 2020). Highly glycolytic tumor cells favor OXPHOS-dependent anti-inflammatory macrophages and compete with glycolytic proinflammatory macrophages.
The metabolism shift of TAMs to glycolysis is regulated by the AKT-mTOR-HIF1α pathway, allowing for macrophage adaption for survival in the hypoxic TME. The hypoxia inducible factor (HIF) family induces reprogramming of pro- to anti-inflammatory macrophages in the TME (Talks et al, 2000). Many glycolysis-related genes such as GLUT1, HK2, PFKFB3, and PGK1 are regulated by HIF1α, leading to preferential glycolytic metabolism of TAMs in hypoxic areas of tumors (Semenza et al, 1994).
Macrophages produce reactive nitrogen and oxygen intermediates at the onset of inflammatory-induced cancers, leading to their genetic instability and malignant transformation (Biswas, 2015). However, macrophages display a proinflammatory status in the early stage of tumor progression and shift their function to an anti-inflammatory phenotype in the late stage of tumor progression.
Metabolic reprogramming significantly affects T cell fate and function (Patsoukis et al, 2016). Each T cell subtype relies on distinct metabolic pathways (Fig. 3). Activated T cells depend on glycolysis and glutaminolysis. Mechanically, the metabolism shift of T cells was mediated by PI3K-AKT-mTOR and their downstream HIF1α and c-Myc (Siska and Rathmell, 2015). Depletion of nutrients by the high glycolytic and glutamine-depleted TME outcompetes and exhausts T cells.
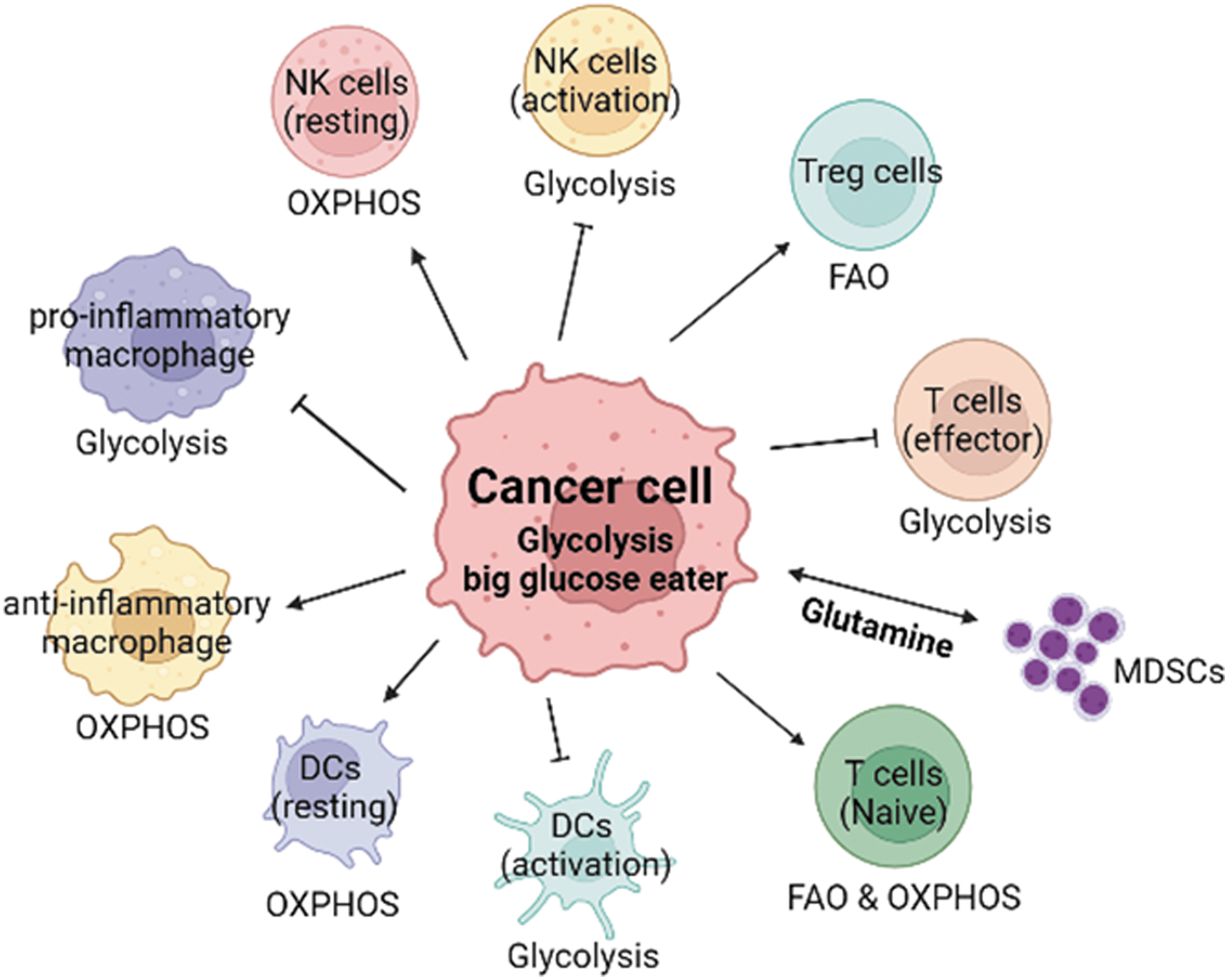
Different immune cell populations rely on distinct metabolic pathways in the TME. Cancer cells, as the biggest glucose consumers, in the TME victimize other glycolytic immune cells, which are tumor-killing cells. Hence, the TME competes with antitumor immune cells in favor of the immunosuppressive milieu. Glutamine metabolism in certain types of cancers can also affect MDSC function in the TME. MDSCs, myeloid-derived suppressor cells.
However, unlike cytotoxic T cells, Tregs predominate the TME and Treg cells mainly rely on fatty acid oxidation rather than glycolysis, hence they are advantageous in the highly glycolytic TME (Macintyre et al, 2014; Michalek et al, 2011). DCs utilize OXPHOS under a resting state, while a shift to glycolysis occurs upon activation. Deteriorated DCs are attributed to decreased glycolysis, but increased lipid storage in the TME (Herber et al, 2010), accompanied by increased expression of immunosuppressive genes ARG1, NOS2, PD-1, PD-L1, and IDO (Biswas, 2015; Tran Janco et al, 2015).
Glycolysis serves as a core metabolic regulator of DC activation, which is regulated by NOS2-induced NO production and activation of the PI3K-AKT pathway. Toll-like receptors (TLRs) are important for the metabolic reprogramming of macrophages, DCs, and T cells in the TME (Huang et al, 2018; Krawczyk et al, 2010). Stimulation of TLRs provokes a metabolic switch from OXPHOS to glycolysis both in macrophages and DCs, ergo supporting proinflammatory macrophages and mature immunogenic DCs (Bullock and Dong, 2014; Kelly and O'Neill, 2015).
TLR8 signaling inhibits tumor cell metabolism and downregulates tumor-derived cyclic adenosine kinase (cAMP) levels, which can rescue senescent naïve and effector T cells (Ye et al, 2014). Recent studies have documented that PD-1 serves as a central regulator of T cells' “exhaustion” state and mediates metabolic reprogramming of immune cells, including T cells and other innate immune cells (Boussiotis and Patsoukis, 2022). PD-1 signaling reduces glycolysis while promoting fatty acid oxidation of T cells (Patsoukis et al, 2015).
Activation of PD-1 signaling in monocytes leads to impaired glycolysis, phagocytosis, and Burton's tyrosine kinase (BTK) signaling (Qorraj et al, 2017). In addition, tumors recruit massive MDSCs that generate reactive nitrogen and reactive oxygen species (ROS) in the TME to reprogram T cells toward the dysfunctional status (Gabrilovich et al, 2012).
Compared with normal adjacent tissue, tumor-specific endothelial cells express SOX18 and SOX7, while pericytes in the TME were enriched in FOXF2 activity (De Val and Black, 2009; Reyahi et al, 2015). ACTA2 and EGR2 genes are highly expressed in TME fibroblasts compared with those in normal tissues (Sathe et al, 2020). The expression of these tumor-specific genes could enhance the identification and functional characterization of TME components. In the TME, fibroblasts produce lactic acid, amino acids, and ketone bodies, which can support tumor cell proliferation. Meanwhile, the glycolytic metabolism of fibroblasts is maintained by cancer cells through the ROS/HIF1α axis (Ghesquiere et al, 2014).
Immune Cell Reprogramming in Circulation Monitors Tumor Progression
Reprogramming of immune cells is localized not only in the TME but also at the systemic level. Tumors alter the organization of immune cells within the body, the extent of which depends on the type of tumor. Changes in immune composition occur in lymph nodes, peripheral blood, bone marrow, and the spleen (Allen et al, 2020). Cancer leads to systemic immune dysfunction, which contributes to tumor progression and closely relates to the patient survival rate and prognosis.
Tumor cells secrete immunosuppressive cytokines, such as TGFβ and IL-10, to reprogram immune cells in the circulation system. Patients suffering from general epithelial malignancy showed an increased number of Tregs (CD4+CD25high) in peripheral blood. Moreover, CD4+ T cells from peripheral blood of cancer patients impaired proliferative capacity (Ormandy et al, 2005; Wolf et al, 2003). In glioblastoma patients, significant elevation of MDSCs, but not Tregs, was found in peripheral blood (Alban et al, 2018).
The systemic-level changes of immune cells are predominantly reported in breast cancer and colorectal cancer. In breast cancer patients, BDCA-1+ DCs and nonclassical monocytes showed reduced TNFα production in response to type I IFN. NK cells displayed a significant drop in IFN-γ production in response to stimuli. T cells showed impairment of IL-2, IL-21, and IFN-γ production (Verronèse et al, 2016). The mRNA expression of PD-L1, FOXP3, CD80, CD40, and CD14 in peripheral blood mononuclear cells (PBMC) increased during breast cancer progression (Kawaguchi et al, 2017).
Breast cancer patients showed a dysregulated IFN-γ signaling pathway in peripheral blood monocytes at diagnosis (Wang et al, 2020). T cell ζ-chain expression in peripheral blood was downregulated in comparison with healthy donors. After surgical tumor resection, T cell ζ-chain expression went back to normal and healthy donor levels (Boniface et al, 2012). These observations indicate that peripheral blood T cells are suppressed in breast cancer patients.
In a case–cohort study of 2774 women, a lower percentage of circulating monocytes was related to a higher risk of breast cancer within 1 year of blood collection, implying that immune cell subsets are altered before tumor initiation (Kresovich et al, 2020). The immune composition in the spleen of breast cancer patients increases in myeloid cells such as neutrophils, eosinophils, and monocytes and decreases in lymphocytes such as T and B cells. The increase in myeloid cells and decrease in lymphocytes become more significant with increasing tumor burden.
These observations offer the potential to screen peripheral blood to detect precancerous states. Compared with healthy donors, colorectal cancer patients showed an increased percentage of Tregs, while decreased expression of the natural cytotoxicity receptors, NKp44 and NKp46, on NK cells and NKT-like cells, in peripheral blood, suggesting that reduced functionality of lymphoid cells in peripheral blood could monitor tumor initiation and development (Krijgsman et al, 2019).
Colorectal cancer patients also showed lower PBMC proliferation and reduced TH1 cytokines (IFN-γ and TNFα) compared with healthy donors (Evans et al, 2010). In contrast, downregulated expression of some natural cytotoxicity receptors on NK cells in some cases was due to viral infections in patients or dependent on the immunological state of individuals. In peripheral blood of breast cancer and colorectal cancer patients, the immune checkpoints, including TIM-3, TIGHT, and PD-L1, were upregulated, while LAG-3 expression was downregulated, compared with healthy donors.
Moreover, the PD-1 promoter was significantly hypermethylated, while PD-L1 was hypomethylated, in breast cancer and colorectal cancer patients, explaining the increased expression level of PD-L1 in cancer cells (Elashi et al, 2019). Compared with healthy donors, PD-1 expression on CD14high myelomonocytic cells, effector T cells, and NK cells showed increasing trends in most renal cell carcinoma patients. PD-1 expression on leukocytes also positively correlated with the cancer progression stage.
After surgical resection of primary tumors, the levels of PD-1 expression on leukocytes decreased significantly (MacFarlane et al, 2014). These observations indicate that the increased expression of immune checkpoint molecules in peripheral blood could be used to monitor the progression of cancer. For hematological malignancies, epigenetic reprogramming driven by CCR6 and KLRG1 caused higher expression of PD-1 on CD8+ T cells within chronic lymphocytic leukemia patients (Wu et al, 2016).
In peripheral blood of acute myeloid leukemia (AML) patients, NK dysfunction supported by NKG2D and NKP30 downregulation and increased population of Treg cells with higher proliferating status have been discovered (Wang et al, 2005). Both CD4+ and CD8+ T cells displayed senescence and exhaustion features (Tang et al, 2020). Gene expression profiling on T cells from AML patients was significantly different compared with healthy donors, coincident with the impaired capacity to form immune synapses (Le Dieu et al, 2009).
Therapeutic Prospects to Target Reprogramming in the TME
Tumor cells reprogram immune cells in both the TME and peripheral tissues to create an immunosuppressive milieu. Comparison of immune cell profiles between cancer patients and healthy donors at transcriptional, metabolic, and functional levels is crucial to inventing therapeutics. A better combination of targeted therapy with conventional cancer therapy will benefit patients.
Metabolic changes in immune cells within the TME affect the function of immune cells. These metabolic regulators, including HIFα, lactic acid, c-Myc, AMP-activated protein kinase (AMPK), and mammalian Target of Rapamycin (mTOR), are being tested for antitumor therapeutics. Metformin, which serves as the first-line medication for type 2 diabetes, increases CD8+ T cell infiltration and protects these CD8+ T cells from apoptosis and exhaustion (Eikawa et al, 2015). The canonical immune checkpoint molecules, PD-1/PD-L1, also play a significant role in reprogramming of T cell metabolism (Patsoukis et al, 2015), supporting the theory that anti-PD-1 immunotherapy might, in part, work through a metabolic change of the TME (Bader et al, 2020).
Tumor cells impair cytotoxic T cells' function through the PD-1/PD-L1 axis. Double blockage antibodies targeting CD47 and PD-L1 (aCD47/aPD-L1) can reprogram the immunosuppressive TME and significantly enhance the antitumor effect of aPD-L1 against primary cancer and metastasis (Hei et al, 2022). PD-L1 knockdown together with OX40L expression can elevate CD4+ and CD8+ infiltration toward the tumor and enhance the adjuvant effect on B cells and DCs (Walters et al, 2021).
Moreover, dietary protein restriction can induce macrophages into a proinflammatory state and inhibit prostate and renal cancer cell proliferation through an ROS/mTOR-centric cascade (Orillion et al, 2018). Dietary fiber and gut microbiota shaped the TME partly through monocyte reprogramming by stimulator-of-interferon-genes-mediated (STING-mediated) IFN signaling (Lam et al, 2021). A glycosylation gene GFPT2 expressed in CAFs controls metabolic reprogramming with increased glucose uptake in lung cancer.
Inhibiting GFPT2 reduced tumor-associated metabolic reprogramming (Zhang et al, 2018). CCL2 facilitates tumor progression in gastric cancer by suppressing macrophages and DCs. Knockout of CCL2 reduced proliferation and metastasis of the tumor through macrophages and NK cell activation (Jeong et al, 2021; Rogic et al, 2021; Yoshimura et al, 2013). CLEVER-1 is upregulated in human breast and head and neck cancers, which promotes the proliferation of tumors and polarization of anti-inflammatory macrophages (Hollmen et al, 2020).
Humanized anti-CLEVER-1 antibody treatment can reinvigorate T cells, NK cells, and peripheral monocytes into an immune activation state. Meanwhile, this reinvigoration endows the peripheral T cells with tumor-killing capacity in several solid cancer patients (Virtakoivu et al, 2021).
Engineering immune cells for therapeutic potential has been a long-standing aspiration of the scientific community, especially for diseases such as cancer and immunodeficiency disorders (Klichinsky et al, 2020). Transducing immune cells with the chimeric antigen receptor (CAR) construct program the immune cell to target cancer cells by recognizing the cancer antigen (Larson and Maus, 2021). Currently, CAR-T, CAR-NK, and CAR macrophages are under clinical trials.
Reinvigoration of suppressed myeloid and lymphoid cells within the TME could increase the efficacy of these CAR-based therapies (Datta et al, 2019). To overcome the cytotoxic side effect and low rate of remission in solid tumors, novel technologies such as mRNA vaccines and nanotherapeutics are promising approaches to enhance the functionality of immune cells in vivo.
Engineering monocytes, DCs, and T cells by mRNA vaccines in combination with CAR technology would increase the efficacy of tumor killing (Reinhard et al, 2020; Rurik et al, 2022; Stephan, 2021).
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The authors received no funding support for this article.