
Abstract
The tumor microenvironment (TME) plays a crucial role in tumor initiation, growth and metastasis. Metabolic enzymes involved in tumor glycolytic reprogramming, including hexokinase, pyruvate kinase, and lactate dehydrogenase, not only play key roles in tumorigenesis and maintaining tumor cell survival, but also take part in the modulation of the TME. Many studies have been devoted to the role of key glycolytic enzymes in the TME over the past decades. We summarize the studies on the role of glycolytic enzymes in the TME of these years and found that glycolytic enzymes remodel the TME primarily through regulating immune escape, angiogenesis, and affecting stromal cells and exosomes. Notably, abnormal tumor vascular system, peritumoral stromal cells, and tumor immunosuppressive microenvironment are important contributors to the failure of antitumor therapy. Therefore, we discuss the mechanisms of regulation by key glycolytic enzymes that may contribute to a promising biomarker for therapeutic intervention. We argue that targeting key glycolytic enzymes in combination with antiprogrammed cell death ligand 1 or antivascular endothelial growth factor could emerge as the more integrated and comprehensive antitumor treatment strategy.
Introduction
The tumor microenvironment (TME) is a special biological environment composed of tumor cells together with infiltrating immune cells, stromal cells, blood vessels, extracellular matrix, and cytokines. In the early stage of tumor growth, a dynamic interrelationship is formed between cancer cells and components of the TME that support cancer cell survival, local invasion, and metastatic spread, thus exerting a pivotal influence in tumor progression, invasion, and metastasis (Roma-Rodrigues et al., 2019). Tumor immune escape is an important way for tumor cells to evade host cell surveillance and attack, and is closely related to the changes of tumor cells and the TME. Therefore, the tumor immunosuppressive microenvironment largely determines whether tumor cells can successfully achieve immune evasion (Lei et al., 2020).
Cancer-associated fibroblasts (CAFs) are important components of the TME, which are involved in tumorigenesis and metastasis by secreting cytokines and metabolites (Biffi and Tuveson, 2021). It has been found that many malignant tumors such as prostate, lung, breast, gastric, colorectal, and pancreatic cancers contain large amounts of CAFs, especially in breast and pancreatic cancers, even up to 80% of the tumor mass (Eisenberg et al., 2020). The interaction between cancer cells and surrounding CAFs significantly impacts the development of cancer, which called “the reverse Warburg effect” (Pavlides et al., 2009). The tumor vascular system and exosomes also play a vital part in the interaction of cancer cells with other cellular in the TME (Anderson and Simon, 2020). However, the current understanding of the characteristics of the interaction between tumor cells and the TME, especially the functions fulfilled by glycolytic enzymes in it, is insufficient and worth further investigation.
Proliferating tumor cells consume large amounts of glucose for glycolytic metabolism rather than aerobic oxidation even under abundant oxygen supply, and produce large amounts of lactic acid, which is known as “the Warburg effect.” This metabolic reprogramming not only provides energy for tumor cell proliferation and metastasis but also affects the TME (Liberti and Locasale, 2016). In recent years, many studies have reported that inhibition of the glycolytic process can alter the activity of metabolites and their component cells in the TME, resulting in inhibition of tumor growth (Bader et al., 2020).
Glycolysis is predominantly mediated by several key enzymes: (1) hexokinase 2 (HK2), which catalyzes the conversion of glucose to glucose 6-phosphate; (2) phosphofructokinase 1 (PFK1), which catalyzes the conversion of glucose 6-phosphate to fructose 6-phosphate; (3) pyruvate kinase M2 (PKM2), which catalyzes the conversion of phosphoenolpyruvate to pyruvate and ATP; and (4) lactate dehydrogenase A (LDHA), which catalyzes the conversion of pyruvate to lactate (Fig. 1). Therefore, exploring the role of key glycolytic enzymes in the remodeling of the TME can deepen our understanding of the mechanisms of tumorigenesis driven by the TME and contribute to the development of promising cancer therapies, improve the effectiveness of tumor treatment, and identify potential biomarkers. This review elucidates the functions of key glycolytic enzymes in the TME and provides a more integrated and comprehensive glycolytic key metabolic enzyme targets for antitumor therapeutic strategies.
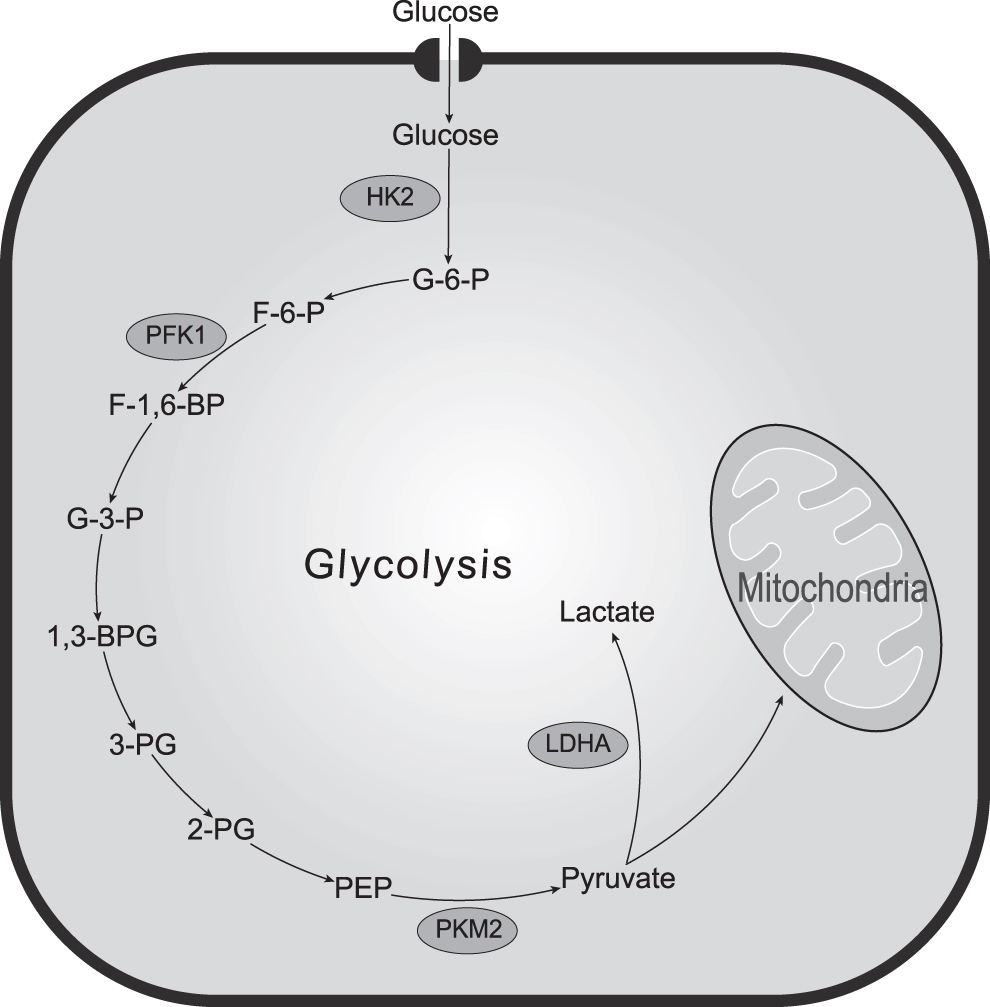
Key Glycolytic enzymes in the cell. HK2: hexokinase 2; G-6-P: glucose-6-phosphate; PFK1: phosphofructokinase 1; F-6-P: fructose-6-phosphate; F-1,6-BP: fructose-1,6-bisphosphate; 1,3-BPG: 1,3-bisphosphoglycerate; 2-PG: 2-phophoglycerate; 3-PG: 3-phosphoglycerate; PKM2: pyruvate kinase 2; PEP: phosphoenolpyruvate.
Key Glycolytic Enzymes Mediate Tumor Immune Escape
Key glycolytic enzymes regulate immune cell activation and infiltration
Immune cells, the key component of the TME, are strongly influenced by metabolic reprogramming. As the first rate-limiting enzyme of glycolysis, HK2 is closely associated with the activation of the inflammatory program in immune cells (Gerriets et al., 2015) and the tumorigenic program in cancer cells (Ho et al., 2015). Previous studies have shown that HK2 expression is highly correlated with the level of immune cell infiltration and immune marker gene expression in renal cell carcinoma (Liu et al., 2022), esophageal cancer (Liu et al., 2021), and glioma (Huang et al., 2022). Overexpression of HK2 in melanoma cells suppresses CD4+ T cell-mediated immune surveillance. Upregulated HK2 in hepatocellular carcinoma (HCC) can inhibit innate immune responses and blunt natural killer (NK) cell sensitivity (Perrin-Cocon et al., 2021).
In addition, PFK1 is involved in macrophage differentiation (Kelly and O'Neill, 2015) and Th17 cell differentiation (Araujo et al., 2017) as the second rate-limiting enzyme of glycolysis. Its isoenzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) also regulates the secretion of C-X-C motif chemokine ligand 12 (CXCL2) and C-X-C motif chemokine ligand 8 (CXCL8) by monocytes through the nuclear factor kappa B (NF-кB) signaling pathway to induce neutrophil recruitment and promote HCC metastasis (Peng et al., 2020). Of note, PKM2 enhances oxidative phosphorylation and Warburg effect to form a dynamic link between energy metabolism and tumor immunity. Upregulated PKM2 in HCC increases the number of CD8+ T cells, Treg cells, and M2 macrophages and suppresses M1 macrophages, which promotes the formation of the immunosuppressive microenvironment (Li et al., 2020b).
Meanwhile, M2 macrophage infiltration correlated with PKM2 expression in pancreatic ductal adenocarcinoma cells, and inhibition of both M2 macrophage polarization, and PKM2 expression in tumor cells could synergistically inhibit tumor growth (Hu et al., 2020). Collectively, most of these key glycolytic enzymes are associated with the activity of immunosuppressive cells such as regulatory T cells and tumor-associated macrophages, but the mechanisms involved have not been fully elucidated and remain to be further investigated.
One of the most significant characteristics of the TME is hypoxia, which leads to increased levels of reactive oxygen species and enhanced tumor invasion (Semenza, 2003). The hypoxia-inducible factor-1α (HIF-α) signaling pathway, regulating multiple enzymes responsible for the glycolytic switch, is identified as an important factor in responding to tumor hypoxic condition (Weidemann and Johnson, 2008). In lung cancer, HIF-1α regulates the expression of several glycolytic enzyme isoforms, including HK2, PFK1, and PKM2 (Li et al., 2020a). In gastric cancer cells, HIF-α enhances LDHA expression by downregulating the transcription factor forkhead box O4 (FOXO4) that binds to the LDHA promoter, thereby increasing the rate of glycolysis in tumor cells (Wang et al., 2021b).
It is noteworthy that excessive lactate, the primary product of LDHA in tumor cells glycolysis, plays an important role in remodeling immune cell function, which facilitates the establishment of tumor immunosuppression (Hayes et al., 2021). For example, lactate inhibits T cell receptor-triggered interferon-gamma, tumor necrosis factor-alpha, and interleukin-2 production and impairs the function of cytotoxic T lymphocyte cells by inhibiting the phosphorylation of p38 signaling protein (Mendler et al., 2012). Lactate can also regulate CD4+ T cell polarization and reduce the percentage of T helper 1 subsets by inducing SIRT1-mediated deacetylation/degradation of T-bet transcription factors (Comito et al., 2019). Therefore, inhibition of lactate anabolism in tumor cells by targeting LDHA has emerged as a potential marker for enhancing the antitumor immune response.
Seth et al. demonstrated in a mouse lung cancer model that knockdown of LDHA in macrophages limits immune escape over time by increasing CD3+ T cells, activating CD8+ T cells, and inducing Th17 and Th1 (Seth et al., 2017), which is consistent with the findings in the mouse HCC experiments (Serra et al., 2022). LDHA could also promote tumor metastasis by upregulating the HIF-1α signaling pathway and inhibiting CD3+ and CD4+ T cell infiltration in the TME (Serganova et al., 2018). Crane et al. found that glioblastoma multiforme secretes LDH5, consisting of the LDHA subunit, to induce the expression of natural killer group 2, member D (NKG2D) ligand in bone marrow cells and monocytes, which inhibits the activation of NK cells bearing the NKG2D receptor to promote immune escape (Crane et al., 2014).
To conclude, the above studies reveal how LDHA affects immune cell-mediated immune escape, which inspires the mechanism of other glycolytic enzymes regulating immune cells, and presents a promising target for immunotherapy of various cancers.
Key glycolytic enzymes regulate protein programmed cell death ligand 1 expression
Programmed cell death ligand 1 (PD-L1) on tumor cell membranes binding to PD-1, which inhibits T cell activation and recruitment, is a critical mechanism for tumor cells to evade immunotherapy (Sharma et al., 2017). It has been analyzed that HK is positively correlated with PD-L1 expression. In mammals, four HK isozymes have been identified, HK1 through HK4. In glioblastoma, high glucose-induced phosphorylation of IкBα by HK2 upregulated NF-кB-associated PD-L1 expression and inhibited CD8+ T cell recruitment, which in turn promoted tumor immune escape (Guo et al., 2022).
Furthermore, HK3 in renal cell carcinoma can impact tumor immune evasion via promoting monocyte and macrophage infiltration and regulating the immunodetectable markers PD-1 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) in exhaustive T cells (Xu et al., 2021). Meanwhile, HK3 demonstrated good efficiency in predicting the efficacy of PD-1 monoclonal antibody in nonsmall cell lung cancer, which provides a potential molecular marker for predicting prognosis (Tuo et al., 2020).
It has been shown that PKM2 and PD-L1 expression is closely related in lung adenocarcinoma, and those with high expression of both tend to have a poorer prognosis (Long et al., 2020). Dyck et al. demonstrated that dimeric PKM2, which binds to the hypoxia response elements of the PD-L1 promoter with HIF-1α in the nucleus, is essential for PD-L1 expression in the mouse colon cancer model (Palsson-McDermott et al., 2017). In HCC, EGF induced translocation of phosphorylated PKM2 into the nucleus to phosphorylate histone H3 at the Thr11 site, which upregulated the expression level of PD-L1 (Wang et al., 2020).
In addition, overexpression of PFKFB3 activates NF-кB to induce PD-L1 expression on peritumoral monocytes, which interferes with CD8+ effector T cells to eliminate HCC cells, causing immune tolerance (Chen et al., 2019). Given the significant association of PKM2 with multiple cancers, we speculate that combination therapy targeting PKM2 and anti-PD-1 antibodies may be a potential antitumor treatment regime to avoid immune escape.
Key Glycolytic Enzymes Mediate Angiogenesis by Regulating Endothelial Cell Activity and Vascular Endothelial Growth Factor Secretion
Tumor blood vessels are vital channels for the source and delivery of tumor nutrients, and tumor angiogenesis plays an important role in promoting tumor development. Regulation of endothelial cells is an influential way for metabolic enzymes to participate in tumor angiogenesis. For example, dimeric PKM2 secreted by cancer cells enters the circulation, which promotes endothelial cell migration and adhesion of endothelial cells to the extracellular matrix (Li et al., 2014). PKM2 in endothelial cells promotes angiogenesis by providing ATP for VE-calmodulin internalization and endothelial cell attachment, whereas it is not involved in endothelial cell proliferation (Gómez-Escudero et al., 2019).
Besides, HK2 is closely associated with endothelial cell apoptosis, which enhances angiogenesis in melanoma by upregulating glycolysis and activating the P38-MAPK signaling pathway (Lu et al., 2019). Vascular endothelial growth factor (VEGF), the key biomolecule in the regulation of angiogenesis, can also be modulated by key enzymes of glycolysis. It has been reported that under hypoxic conditions in pancreatic cancer cells, PKM2 overexpressed in the cytoplasm translocate into the nucleus to bind to NF-кB and activate HIF-1α and its target gene VEGF, leading to increased VEGF secretion and contributing to tumor angiogenesis (Azoitei et al., 2016). Moreover, PKM2 overexpressed in the cytoplasm of bladder cancer cells binds to STAT3 to form a complex to translocate into the nucleus to activate HIF-1α and VEGF (Xia et al., 2022). Knockdown of PFKFB3 in breast cancer cells could downregulate VEGFα protein expression and repressed angiogenic activity (Peng et al., 2018).
Tumor vascular normalization is recently emerging as a new approach to improve the efficacy of antitumor therapy. Cantelmo et al. identified that reducing VE-calmodulin endocytosis in endothelial cells through inhibition of PFKFB3 could promote tumor vascular normalization and enhance the vascular barrier, hence improving tumor vascular perfusion and inhibiting tumor metastasis (Cantelmo et al., 2016), which is consistent with the results in the mouse HCC model (Matsumoto et al., 2021). However, the use of high doses of PFKFB3 inhibitor 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) can instead inhibit endothelial cell growth and disintegrate tumor blood vessels, leading to the destruction of the vascular barrier to promote cancer cell invasion and metastasis (Conradi et al., 2017). It implies that appropriate doses of glycolysis inhibitors may be more beneficial.
In a mouse model of glioblastoma, dual inhibition of PFKFB2 and VEGF was proven to improve tumor vascular function, remodel the TME, and enhance chemotherapeutic efficacy via suppressing the expression of Tie 1, a key regulator of tumor vascular normalization (Zhang et al., 2020). Given the effect of PFKFB2 on tumor vascular normalization, anti-VEGF combined with targeting PFKFB3 is a very promising tumor vascular normalization regimen, which creates a window of opportunity to improve the efficacy of chemotherapy.
Key Glycolytic Enzymes Mediate the Reverse Warburg Effect of CAFs
The interaction between CAFs and tumor cells, which crosstalk and undergo metabolic reprogramming, contributes to the activation of CAFs and the growth of tumor cells. Specifically, cancer cells secrete hydrogen peroxide into the TME to induce oxidative stress in neighboring stromal cells, CAFs undergo aerobic glycolysis and produce high levels of energy-rich metabolites (e.g., pyruvate, lactate, fatty acids) for delivery to tumor cells. The mitochondrial oxidative phosphorylation system produces large amounts of ATP, which allows tumor cells to have a higher proliferative capacity (Pavlides et al., 2009). In fact, the key glycolytic enzymes of CAFs play an instrumental part in the reverse Warburg effect. It was observed that upregulation of PK expression in CAFs could induce CAFs to produce large amounts of lactate and ketone bodies to support tumor growth (Chiavarina et al., 2011).
Oncogenic gene c-Myc enhances CAFs glycolysis and increases glucose uptake in tumor cells via upregulation of LDH and glucose transporter 1 expression (Sun et al., 2014). In chronic hypoxic conditions created by tumors, CAFs tend to convert to a pro-glycolytic phenotype and overexpress LDH and PKM. Therefore, targeting key glycolytic enzymes becomes an inevitable strategy to impede this effect. Using glycolysis inhibitors in breast cancer lacking stromal caveolin-1 significantly inhibited tumor growth induced by CAFs overexpressing PKM2 and LDH (Bonuccelli et al., 2010). Inhibition of LDH in lung cancer cells can hinder their metabolic cooperation with CAFs and inhibit cancer cell migration (Koukourakis et al., 2017). Most of the above-mentioned studies illustrate that key glycolytic enzymes play a facilitative role in the procarcinogenic effects of CAFs, but do not explain the relevant mechanisms involved, and there is still room for research on how glycolytic enzymes affect CAFs.
Key Glycolytic Enzymes Increase Extracellular Vesicle Biogenesis
Extracellular vesicles (EVs) are a heterogeneous set of cell-derived membrane structures, which mediate intercellular communication of great significance in tumor development (Meehan and Vella, 2016). According to the biological properties or release pathways, EVs can be classified into microvesicles and exosomes, which originate from the endosomal system or which are shed from the plasma membrane, respectively (van Niel et al., 2018). The extracellular secretion of tumor cell exosomes is mainly regulated by the syndecan-syntenin-ALIX pathway (Baietti et al., 2012) and the soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor (SNARE) complex, which consists of proteins on the cell membrane (t-SNARE) and on the membrane of the outgrowing vesicles (v-SNAREs), including synaptosome-associated protein 23 (SNAP-23) (Guo et al., 1998).
During exosome secretion, PKM2 forms a dimeric structure with high protein kinase activity and then binds to SNAP-23 near the cell membrane, leading to phosphorylation of SNAP-23 at the Ser95 site and upregulating exosome release from tumor cells (Wei et al., 2017). Consequently, PKM2 mainly exerts its noncanonical function to regulate exosome secretion instead of its metabolic function.
Exosome-associated glycolytic enzymes also have impact on the process of tumor drug resistance and development. Hypoxia-induced exosome-associated PKM2 can deliver cisplatin resistance to sensitive nonsmall cell lung cancer cells, and by applying Shikonin, this resistance effect can be blocked and the efficiency of cisplatin treatment can be improved (Dai et al., 2022, Wang et al., 2021a). In addition, prostate cancer cells promote the production of CAFs-derived CXCL12 by secreting exosome-associated PKM2 into bone marrow stromal cells to induce HIF-1α expression, which promotes the seeding and growth of prostate cancer cells in the bone marrow by binding to the C-X-C motif chemokine receptor 4 (CXCR4) on prostate cancer cells (Dai et al., 2019). It makes exosome-associated PKM2 available as a potential biomarker for the treatment of prostate cancer bone metastases.
Ectosomes are also EVs, which shed directly from the plasma membrane. Although there are significant differences between exosomes and ectosomes, their functions are thought to be broadly similar. The plasma membrane-localized protein arrestin-domain-containing protein 1 (ARRDC1) regulates protein cargo and release of EVs (Nabhan et al., 2012). In HCC cells, sumoylated PKM2 was enriched in ectosomes as well as ectosomally excreted by interaction with ARRDC1. HCC-derived ectosomal PKM2, which is transmitted into monocytes, induces polarization of monocytes into M2-like macrophages through phosphorylating STAT3 in the nucleus. Macrophage-secreted chemokine C-C motif ligand 1 (CCL1) binds its receptor C-C motif receptor 8 (CCR8) in HCC cells and induces AKT activation-enhanced interaction between PKM2 and ARRDC1, further facilitating PKM2 excretion from HCC cells to form a feedforward regulatory loop for tumorigenesis (Hou et al., 2020).
In summary, PKM2 in tumor cells promotes the secretion of EVs, and induces transcriptional reprogramming in recipient cells, through their noncanonical functions and activation of transcription factors such as HIF-1α and STAT3. Transcriptionally reprogrammed cells differentiate into subtypes that favor tumor growth and secrete factors or metabolites that promote tumor cell growth and metastasis. Whether other glycolytic enzymes are also involved in influencing EVs secretion deserves to be investigated.
Conclusion
With the development of research on the TME, a large number of articles have elucidated the roles of immune cells, stromal cells, and tumor vascular system in the TME, as well as new potential targets for antitumor therapy. Metabolic enzymes, as direct drivers of aberrant tumor metabolism, especially key glycolytic enzymes, are not only involved in regulating the activities of tumor cells, but also act in remodeling the TME. We summarize that key glycolytic enzymes remodel the TME in the following ways: (1) constructing an immune escape environment by inducing differentiation and recruitment of immunosuppressive cells, and regulating PD-L1 expression; (2) promoting endothelial cell migration and VEGF secretion to mediate tumor angiogenesis; (3) mediating the reverse Warburg effect of CAFs to provide nutritious environment for tumor proliferation; and (4) facilitating exosome secretion and reprogramming the recipient cells to differentiate into subtypes favorable for tumor growth (Fig. 2).

Functions of key glycolytic enzymes in the TME. Key glycolytic enzymes regulate the TME in several ways: (1) mediating immune escape through inducing proliferation and differentiation of immunosuppressive cells, suppressing immune effector cells, and regulating PD-L1 expression; (2) promoting endothelial cell migration and VEGF secretion to regulate tumor angiogenesis; (3) mediating the reverse Warburg effect of CAFs and providing energy-rich metabolites for tumor proliferation; and (4) promoting the secretion of EVs, and reprogramming recipient cells to differentiate into subtypes favorable for tumor growth. CAFs, cancer-associated fibroblasts; EVs, extracellular vesicles; PD-L1, programmed cell death ligand 1; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
Key glycolytic enzymes have found to be important contributors to tumorigenesis, invasion, and metastasis, and themselves provide valuable therapeutic targets for the treatment of tumors. However, this development process is still challenging. Tumor cells will adopt different metabolic regimens due to differences in the TME, which makes tumor metabolic mechanisms often highly heterogeneous. At the same time, the complex network of cellular metabolism is highly compensatory, making it difficult to achieve long-lasting effects with any single target intervention. Therefore, we assume that targeting key glycolytic enzymes in combination with other antitumor therapies such as anti-PD-L1 and anti-VEGF may emerge as a promising direction for future studies and provide a more integrated and comprehensive antitumor treatment strategy.
Footnotes
Authors' Contributions
C.Z. had the idea for the article. W.X. and J.W. wrote the article. M.X. and S.L. performed the literature search and data analysis. N.R., Q.Z., and Z.H. designed figures. Y.S. and C.Z. supervised all project and critically revised work. All authors edited and reviewed the content of the article and approved the final article.
Consent for Publication
All authors consent to the publication of this study.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was supported by the National Natural Science Foundation of China (82103521, 82172799, 82073208), the Special Foundation for Science and Technology Basic Research Program (2019FY101103), the Shanghai Sailing Program (21YF1407500), China Postdoctoral Science Foundation (2021M690674), and the Shanghai Shen Kang Hospital Development Center New Frontier Technology Joint Project (SHDC12021109).