
Abstract
Cord blood (CB) is widely stored as a source of hematopoietic stem cells for potential future use, though its application for autologous purposes remains limited. Repurposing CB into human-induced pluripotent stem cells (hiPSCs) can broaden its utility beyond hematological conditions. This study investigated the effects of umbilical cord-mesenchymal stromal cell (UC-MSC) co-culture on CB CD34+ cells and the characteristics of the resulting hiPSCs. CD34+ cells were isolated, expanded in UC-MSC co-culture for 3 days, and reprogrammed into hiPSCs using episomal vectors. Results showed that UC-MSC co-culture significantly increased CD34+ cell numbers (p < 0.0001, n = 6), with a reduced population doubling time of 25.1 ± 2.1 hours compared with the control (p < 0.0004, n = 6). The yield of CD34+ cells was substantially higher in the UC-MSC co-culture group. The hiPSCs exhibited comparable reprogramming efficiency, pluripotency marker expression, trilineage differentiation potential, and genomic stability to CD34+ cells expanded under standard culture conditions. These findings suggest that CD34+ cells from CB, expanded in UC-MSC co-culture, can be reprogrammed into functional hiPSCs without compromising cell quality or genetic stability.
Introduction
Cord blood (CB) is a valuable source of hematopoietic stem cells (HSCs) due to its youthful and relatively disease-free nature, which reduces the risk of pathogen transmission compared with other adult cell sources. The practice of cryopreserving CB for potential future use is well-supported, with over 40,000 successful HSC transplants performed in both children and adults for the treatment of malignant and non-malignant hematological diseases (Mayani et al., 2020). A critical factor in determining whether a donated CB unit is suitable for cryopreservation is the number of CD34+ HSCs. Specifically, a minimum CD34+ cell count of ≥1.5 × 105 cells/kg per single unit or ≥1 × 105 cells/kg per unit for double unit infusion is required; samples failing to meet these thresholds are typically discarded (Dehn et al., 2019; Politikos et al., 2020; Rocha et al., 2009). Unfortunately, a significant portion of collected CB, ranging from 36% to 39%, is discarded due to insufficient CD34+ cell counts, resulting in considerable economic loss, depletion of resources, and waste of valuable samples, including those from donors with rare blood types or conditions (Tan, 2017). This underscores the urgent need to develop in vitro cell expansion techniques that can increase the yield of CD34+ cells without compromising their quality for cellular reprogramming or therapeutic use.
Cellular reprogramming technology has revolutionized regenerative medicine by enabling the conversion of various somatic cells into pluripotent stem cells, known as the human-induced pluripotent stem cells (hiPSCs). Consequently, repurposing CB through cellular reprogramming to generate hiPSCs holds significant promise for expanding therapeutic applications due to the high differentiation potential of these cells (Abberton et al., 2022; Alvarez-Palomo et al., 2022). In the laboratory, hiPSCs exhibit pluripotent capabilities akin to those of human embryonic stem cells, achieved through the reprogramming of adult somatic cells (Puri and Nagy, 2012; Takahashi et al., 2007; Yu et al., 2007). These pluripotent cells can be differentiated into a variety of cell types, including neurons and cardiomyocytes, or maintained as a continuous source of HSCs, particularly valuable for donors with rare blood types.
However, the low cell content in some CB grafts can limit the number of cells available for reprogramming. Additionally, extensive in vitro expansion of hematopoietic cells has been associated with asymmetrical division, leading to heterogeneous progeny and depletion of the symmetrically dividing HSC pool. Previous studies have demonstrated that the proliferation of CB-derived CD34+ HSCs can be significantly enhanced through co-culture with mesenchymal stromal cells (MSCs) (Bucar et al., 2021; Lau et al., 2017; Robinson et al., 2006). This effect is likely due to paracrine interactions facilitated by direct MSC-HSC contact, which mimics the supportive niche required for HSC expansion (Chang et al., 2021). However, it remains unclear whether such modifications impact subsequent reprogramming and the generation of hiPSCs. In this study, we demonstrate that CD34+ cells isolated from frozen CB can be expanded through umbilical cord (UC)-MSC co-culture and successfully reprogrammed into high-quality, functional hiPSCs.
Materials and Methods
Human samples and ethics approval
Human CB units were provided by CryoCord Sdn Bhd with informed donor consent. The use of this cell source was reviewed and approved by the Universiti Sains Malaysia Human Ethics Committee (USM/JEPeM/19040247). Three frozen CB units (4.5 mL) were provided with anonymous labels, namely, CB023873, CB012604, and CB013104. The information of the cell source and donor identity were blinded by all investigators. Donor-derived primary human Wharton’s jelly-derived MSCs were also provided by CryoCord Sdn Bhd, which was used in our previous study (Ng et al., 2019).
Isolation and enrichment of HSC CD34+ cells from cryopreserved CB
Mononuclear cells (MNCs) were isolated from 18-year cryopreserved CB using a Lymphoprep™ density gradient medium (STEMCELL Technologies, Vancouver, Canada). The CB was diluted with Dulbecco’s Phosphate-Buffered Saline (PBS) and 2% fetal bovine serum (FBS) (Thermo Fisher Scientific, Massachusetts, USA) and layered over the Lymphoprep™ solution in a centrifuge tube, carefully avoiding mixing. After centrifugation at 800 × g for 30 minutes at room temperature, MNCs were isolated and washed with Dulbecco’s Modified Eagle Medium (DMEM)/Nutrient Mixture F-12 supplemented with 2% FBS (Thermo Fisher Scientific, Massachusetts, USA). Red blood cells (RBCs) were removed using Gey’s RBC lysis (Meurer et al., 2016). CD34+ cells were then positively selected using the EasySep™ Human CB CD34 Positive Selection Kit II (STEMCELL Technologies, Vancouver, Canada) and cultured in Hematopoietic Progenitor Expansion Medium XF with cytokine mix E ((PromoCell, Heidelberg, Germany).
The isolation of CD34+ expressing HSCs and UC-MSC co-culture
Primary human Wharton’s jelly-derived MSCs were obtained from CryoCord Sdn Bhd. UC-MSCs were cultured up to passages 4–5 before use. The cells were maintained in DMEM with 10% FBS (Thermo Fisher Scientific, Massachusetts, USA) until they reached 70%–80% confluence. To minimize FBS effects on HSC expansion, FBS was removed during UC-MSC co-culture. CD34+ HSCs (30,000 cells/mL) were added and maintained for 3 days. Total viable cells, as determined using 0.4% Trypan blue cell exclusion dye, were counted at defined time on a standard hemacytometer under a light microscope. CD34+ cell doubling time was calculated using Equation (1), as below:
Equation (1): Calculation of CD34+ cell population doubling time
Cellular reprogramming and generation of hiPSCs
CD34+ HSCs were reprogrammed using episomal vectors encoding OCT4, SOX2, KLF4, L-MYC, and LIN28, along with mutant p53 and Epstein-Barr nuclear antigen-1 (EBNA) vectors from the Episomal iPSC Reprogramming Kit (Thermo Fisher Scientific, Massachusetts, USA). Electroporation of 1.0 × 105 CD34+ HSCs was performed with the Neon Transfection System at 1650 V, 10 ms pulse width, and three pulses. Transfected cells were then cultured overnight on a Geltrex or Matrigel matrix-coated 6-well plate in Hematopoietic Progenitor Expansion Medium with Cytokine Mix E (PromoCell, Heidelberg, Germany). The next day, N2B27 medium (Thermo Fisher Scientific, Massachusetts, USA) was added without removing the existing medium, and refreshed daily from days 2 to 9 post-transfection. On day 10, the medium was switched to Essential 8™ (Thermo Fisher Scientific, Massachusetts, USA) for continued culture. Between days 15 and 21, hiPSC colonies were observed, manually isolated, and transferred to fresh Essential 8™ medium. Transfection efficiency was calculated as below:
Equation (2): Calculation of hiPSC reprogramming efficiency
Immunostaining
Immunostaining was performed to qualitatively assess cell surface marker expression. Cells were fixed with ice-cold methanol (Sigma-Aldrich, St Louis, USA) at 4°C for 20 minutes. Blocking was done with 5% donkey serum (Thermo Fisher Scientific, Massachusetts, USA) for 30 minutes at room temperature. Samples were incubated with the primary antibody diluted in the blocking solution overnight at 4°C. The next day, samples were washed three times with PBS and incubated with the secondary antibody for 1 hour at room temperature. Afterward, the samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI,Thermo Fisher Scientific, Massachusetts, USA) for 15 minutes, and images were captured using a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Flow cytometric analysis
hiPSCs were dissociated with 0.5 mM UltraPure Ethylenediaminetetraacetic acid (EDTA) and centrifuged at 300 × g for 5 minutes. Cells were then fixed using fixation/permeabilization solution per the manufacturer’s instructions. The primary antibody TRA-1-60 (diluted at 1:200) or Stage-specific embryonic antigen-4 (SSEA4, diluted at 1:300) (BioLegend, California, USA) was added and incubated for 1 hour on ice, followed by centrifugation at 250 × g for 5 minutes. The secondary antibody (Alexa Fluor 488, 1:500) was then added and incubated for 30 minutes. After incubation, samples were washed twice with Perm/Wash buffer before flow cytometric analysis using the NovoCyte Flow Cytometer (ACEA Biosciences, California, USA).
Quantitative real-time polymerase chain reaction
Cells were lysed and harvested using the Monarch Total RNA Miniprep Kit and the RNeasy Mini Kit (New England Biolabs, Massachusetts, USA). RNA purity and concentration were assessed using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA). Once RNA with sufficient purity and quantity was obtained, it was converted to cDNA using Quantiscript Reverse Transcriptase (Qiagen, Hilden, Germany) on a Bio-Rad T100™ Thermal Cycler. Quantitative polymerase chain reaction (qPCR) was then performed using the Quantinova SYBR Green Kit (Qiagen, Hilden, Germany), with fluorescence intensity measured on a StepOnePlus™ qPCR machine (Applied Biosystems, California, USA). Gene expression levels were normalized to the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences used were as follows: PODXL (TRA-1-160) Forward: 5ʹ-AGC TAA ACC TAA CAC CAC AAG C-3ʹ, Reverse: 5ʹ-TGA GGG GTC GTC AGA TGT TCT-3ʹ; REX1 (ZFP42) Forward: 5ʹ-AGA AAC GGG CAA AGA CAA GAC-3ʹ, Reverse: 5ʹ-GCT GAC AGG TTC TAT TTC CGC-3ʹ; GAPDH Forward: 5ʹ-TTG AGG TCA ATG AAG GGG TC-3ʹ, Reverse: 5ʹ-GAA GGT GAA GGT CGG AGT CA-3ʹ (Matrioux, Kuala Lumpur, Malaysia).
Karyotype analysis
hiPSCs were cultured in StemMACS™ iPS-Brew XF (Miltenyi Biotec, Bergisch Gladbach, Germany) for 3 days for optimal growth. Colcemid solution (Thermo Fisher Scientific, Massachusetts, USA) was then added at 10 μg/mL and incubated for 2 hours to arrest cells at metaphase. Cells were trypsinized into single-cell suspensions, centrifuged at 1000 rpm for 10 minutes, and resuspended in 0.56% potassium chloride (Thermo Fisher Scientific, Massachusetts, USA) for 20 minutes at 37°C, followed by centrifugation. Cells were fixed with a 3:1 methanol-acid fixative, incubated for 15 minutes, then treated with a 2:1 fixative ratio and refrigerated overnight. The following day, cells were spread onto slides and stored at −20°C for G-banding analysis. Chromosomes were visualized under BX51 Olympus microscope and analyzed using the Cytovision 7.4 (Leica Biosystem Richmond, Illinois, USA).
Transgene detection
hiPSCs were isolated and resuspended in lysis buffer at a concentration of 1 × 105 cells per test. To detect episomal plasmids, the EBNA-1 primer set (Thermo Fisher Scientific, Massachusetts, USA) was used for all five episomal vectors, while the oriP primer set (Thermo Fisher Scientific, Massachusetts, USA) identified all plasmids except pCXB-EBNA1. The specific primers used for amplification were as follows: OriP (PEP4-SF1-oriP: 5ʹ-TTC CAC GAG GGT AGT GAA CC-3ʹ and PEP4-SR1-oriP: 5ʹ-TCG GGG GTG TTA GAG ACA AC-3ʹ, 544 base pair [bp]) and EBNA (PEP4-SF2-oriP: 5ʹ-ATC GTC AAA GCT GCA CAC AG-3ʹ and PEP4-SR2-oriP: 5ʹ-CCC AGG AGT CCC AGT CA-3ʹ, 666 bp). Amplification was performed using a thermal cycler (Thermo Fisher Scientific, Massachusetts, USA), and the presence of the target vectors was confirmed through agarose gel electrophoresis.
Mycoplasma test
hiPSCs (1 × 105 cells) were lysed and resuspended in lysis buffer for DNA extraction using PrimeWay Genomic II DNA Extraction kit (1st BASE, JTC MedTech Hub, Singapore). The extracted DNA was then mixed with the PCR reaction mix from the Universal Mycoplasma Detection Kit (ATCC, Virginia, USA), which targets the 16S rRNA coding region of the mycoplasma genome with specific primers. PCR products were analyzed on a 1% agarose gel to confirm mycoplasma contamination. A distinct band within the range of 434–468 bp indicated contamination, with positive controls showing a band at 464 bp. Negative controls should display no visible bands.
Short tandem repeat analysis
Short tandem repeat (STR) analysis was outsourced to Apical Scientific Malaysia. Briefly, the genomic DNA of hiPSCs (3 × 105 cells) was extracted using PrimeWay Genomic II DNA Extraction kit (1st BASE, JTC MedTech Hub, Singapore) and quantified to ensure the starting material was of high quality and of sufficient quantity. Multiplex PCR was used to amplify targeted loci, including 15 STR loci with the GenePrint® 24 System (Promega, Wisconsin, USA), covering amelogenin and DYS391. The samples were processed with the ABI 3730XL Genetic Analyzer, and data were analyzed with GeneMapper® v5.0 software (Applied Biosystems, California, USA). Positive and negative controls were included to validate results, and percentage matches were calculated using the formula as below:
Equation (3): Calculation of the percentage matched alleles
Cardiomyocyte differentiation
HiPSCs were cultured in StemMACS iPS-Brew XF (Miltenyi Biotech, Rhineland, Germany) on growth factor-reduced Matrigel (Corning Life Sciences, New York, USA) to 80% confluency. Cardiomyocyte differentiation was then initiated by treating the cells with 6 μm CHIR99021 (Tocris Bioscience, Bristol, UK) in Roswell Park Memorial Institute 1640 (RPMI-1640) medium supplemented with B27 (RPMI+B27) minus insulin (Gibco, Massachusetts, USA) for 48 hours. The medium was refreshed and maintained for 24 hours, followed by treatment with 10 μm XAV939 (Tocris Bioscience, Bristol, UK) and 1 μm BMS184594 in RPMI+B27 minus insulin medium for 48 hours. After another 48 hours, the medium was changed to RPMI+B27 plus insulin, refreshed every 2 days. Beating cells, indicative of cardiomyocyte differentiation, typically appeared by day 10. Differentiation was confirmed by immunostaining with 1:500 dilutions of cardiac troponin T (cTnT) and myosin light chain ventricular (Abcam, Cambridge, USA).
Dopaminergic neuron differentiation
Dopaminergic neuron differentiation was performed following the protocol established by Kikuchi et al. (2017), with an optimized concentration of 0.75 μm CHIR99021 (Tocris Bioscience, Bristol, UK). Progenitor cells were generated by day 16 through dual-suppressor of Mothers against Decapentaplegic (SMAD) inhibition (Tocris Bioscience, Bristol, UK). These progenitor cells were re-plated and matured in B27 medium with 20 ng/mL Brain-derived neurotrophic factor (BDNF), 10 ng/mL Glial Cell Line-derived Neurotrophic Factor (GDNF), 200 mM ascorbic acid, and 1 μm Dibutyryl-cyclic adenosine monophosphate (dbcAMP, Sigma Aldrich, Missouri, USA). Dopaminergic neurons typically developed by day 44. Immunostaining for dopaminergic neurons used 1:500 dilutions of beta-III tubulin and tyrosine hydroxylase (TH) (Abcam, Cambridge, USA).
Endodermal differentiation
hiPSCs were cultured in StemMACS iPS-Brew XF (Miltenyi Biotech, Rhineland, Germany) at 20,000 cells/cm2 on growth factor-reduced Matrigel (Corning, New York, USA). After 24 hours, they were transitioned to RPMI-1640 (Gibco, Massachusetts, USA) with 100 ng/mL activin A (Tocris Bioscience, Bristol, UK) and treated with 5 µM CHIR99021 (Tocris Bioscience, Bristol, UK) for 1 day. The medium was then changed to RPMI-1640 with B27 insulin (Gibco, Massachusetts, USA) and 100 ng/mL activin A for 2 days. Endoderm layer emergence, indicating successful differentiation, was observed by day 3. Endodermal differentiation was confirmed by immunostaining with 1:500 dilutions of sex-determining region Y-box 17 (SOX17) and forkhead box protein A2 (FOXA2) (BioLegend, California, USA).
Statistical analysis
The data were expressed as mean ± standard error of the mean from at least three biological replicates (N = 3) or more. All statistical analyses were performed using GraphPad Prism 8.0.2. The data between two groups were analyzed using an unpaired Student’s t-test. Statistical significance was considered when p < 0.05.
Results
UC-MSC co-culture promotes the expansion of CB-derived CD34+ HSCs
MSCs showed fibroblast-like morphology at passage 4 and were grown to 70%–80% confluent prior to the subsequent co-culture experiment (Fig. 1A). An average of 18.35 ± 3.12 × 106 (n = 5) MNCs were isolated from individual 4.5 mL cryopreserved CB samples and with an average cell density of 4.08 ± 0.31 × 106 cells/mL (CB023873, CB012604, and CB013104). Flow cytometry analysis revealed that 2.70% of the MNCs were CD34+ (Fig. 1B). After 24 hours of magnetic sorting, 1.7 ± 0.33 × 106 CD34+ cells were successfully purified (Fig. 1B). Three-day time-lapse imaging of the culture showed a significantly higher number of floating, and mononuclear bridge cells were observed in UC-MSC co-culture (Fig. 1C). The total CD34+ HSCs was also markedly increased from 1.5 ± 0.2 × 105 to 9.7 ± 0.1 × 105 cells in the UC-MSC co-culture as compared with only 2.7 ± 0.3 × 105 cells in the control (p < 0.0001, n = 6, Fig. 1D). These results were further supported by the significant reduction in the population doubling time of CD34+ cells in the UC-MSC co-culture from 103.3 ± 41.2 hours in the control to 25.1 ± 2.1 hours (p < 0.0004, n = 6, Fig. 1E).
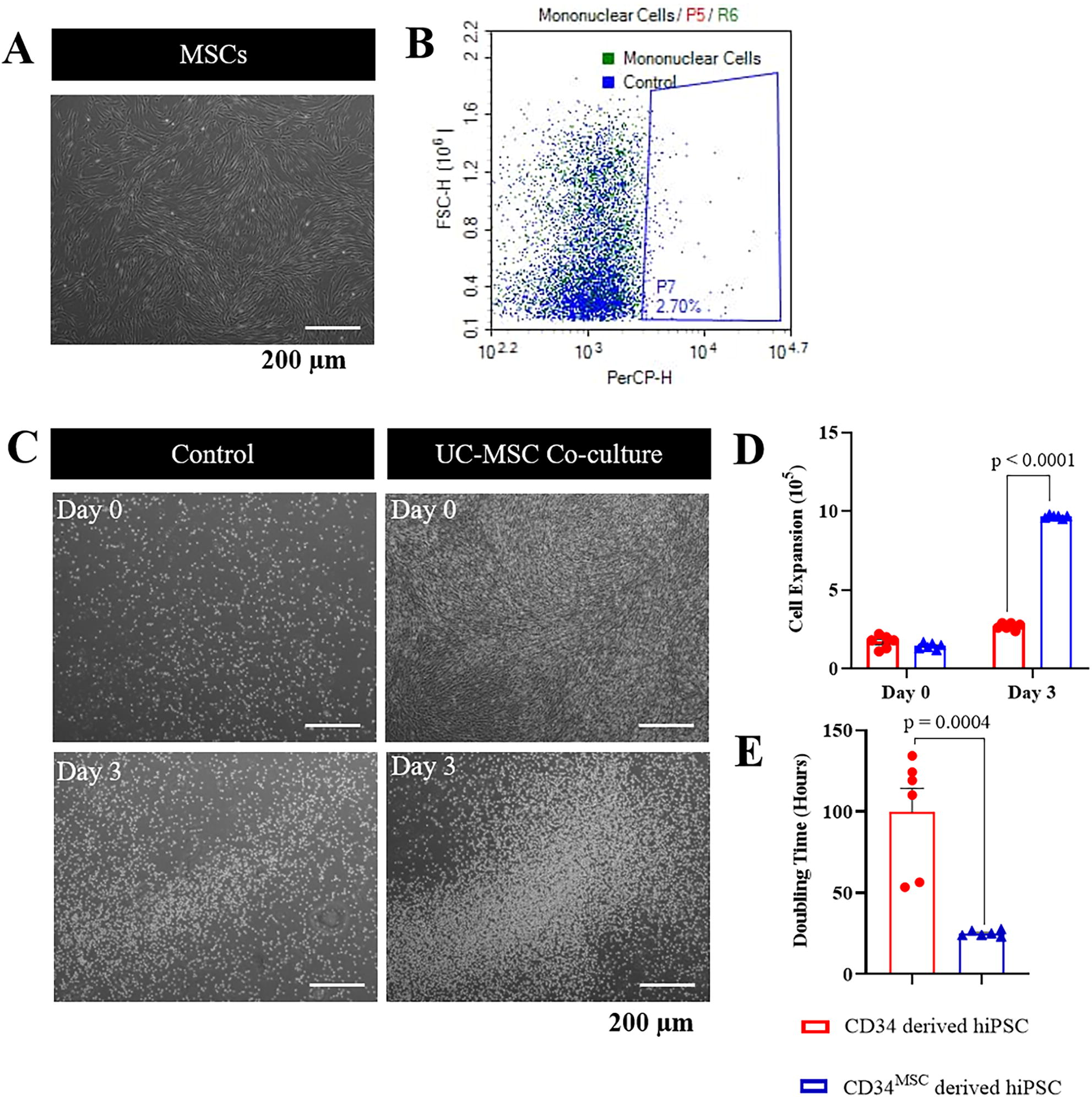
UC-MSC co-culture promotes the expansion of CD34+ HSC CB.
Matrigel promotes growth of early reprogrammed CD34 cells
The transfected cells were observed to transition from suspension, non-adherent bright cells to adherent, fibroblastic-like cells as early as day 5 under standard protocol using Geltrex (Fig. 2A). To optimize the growth of the early reprogrammed iPSCs, the effects of two commercially available, growth factor-reduced basement membrane Matrigel and Geltrex were assessed. Time-lapse imaging of reprogrammed cells showed that early attachment of reprogrammed cells seeded on Matrigel (day 5) than on the Geltrex (day 8, Fig. 2B). However, the average hiPSC colony size was found larger on Geltrex (2.5 ± 0.1 mm2) compared with Matrigel (1.28 ± 0.0 mm2) (Fig. 2C). Despite the difference, there was no significant change to the cell reprogramming efficiency, either the cells were cultured on Matrigel (0.06 ± 0%, n = 3) or Geltrex (0.07 ± 0.002%, n = 3).
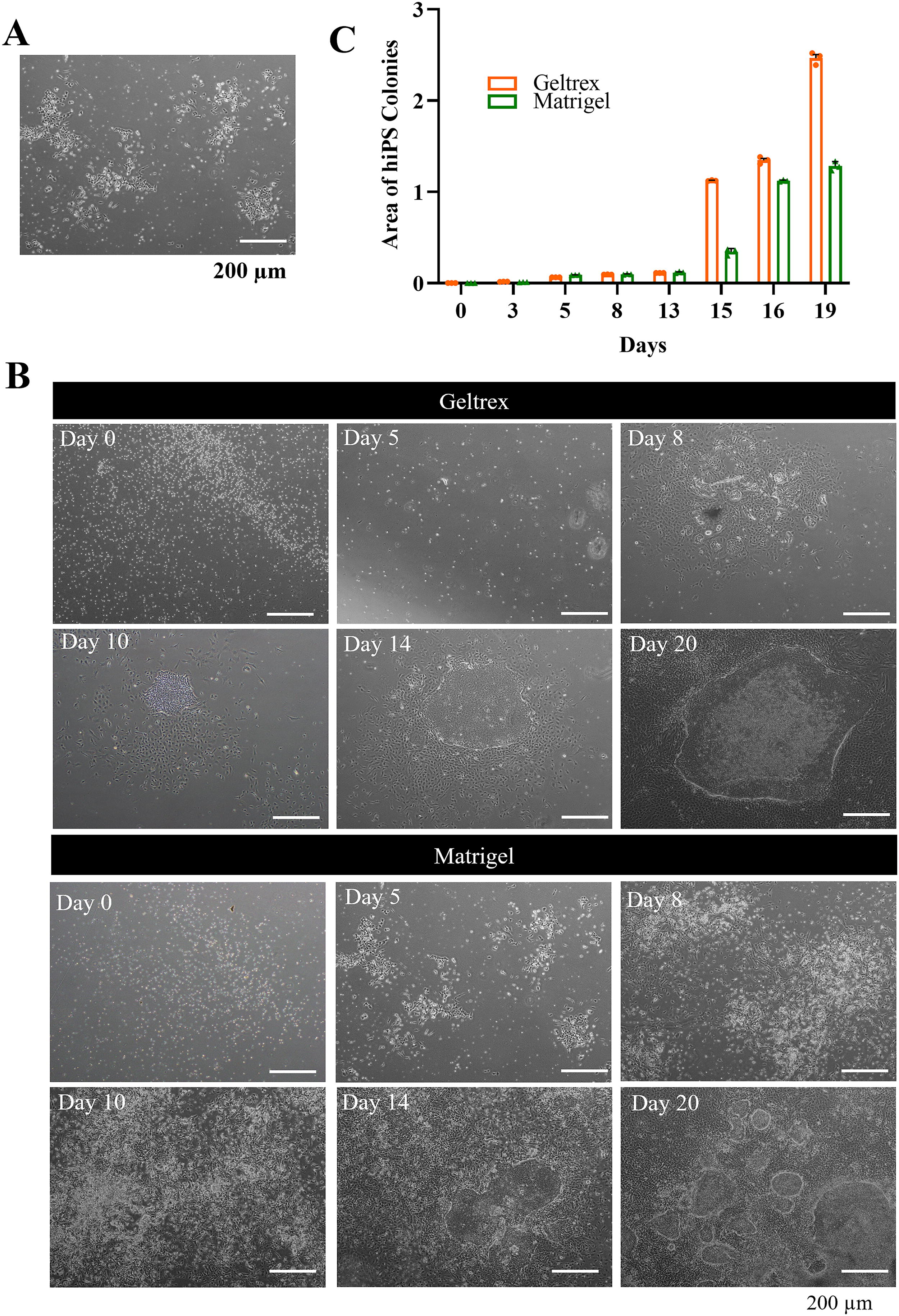
Morphology and colony formation in PromoCell CB-iPSCs.
Co-culture CD34 cells with UC-MSC does not compromise the reprogramming efficiency and pluripotent characteristics of the produced CD34MSC-derived hiPSC
Reprogrammed CD34 hiPSC line derived from UC-MSC co-culture with CD34+ HSCs (0.05 ± 0.0012) did not compromise reprogramming efficiency compared with expanded CD34+ HSCs without co-culture (0.05 ± 0.0015, n = 6, Fig. 3A). The expression of pluripotency markers TRA-1-60 and SSEA4 were expressed in both CD34+-derived hiPSCs and CD34MSC-derived hiPSCs (Fig. 3B). Flow cytometric analysis revealed 94.1 ± 0.6% of CD34+-derived hiPSCs expressed TRA-1-60 and 93.1 ± 0.1% expressed SSEA4; whereas 92.2 ± 0.4% CD34MSC-derived hiPSCs expressed TRA-1-60 and 91.7 ± 0.6% expressed SSEA4 (Fig. 3C). qPCR analysis further confirmed the expression of TRA-1-60 and zinc finger protein 42 (ZFP42) genes in hiPSCs were unaffected by UC-MSC co-culture of the parental CD34 cells (Fig. 3D).
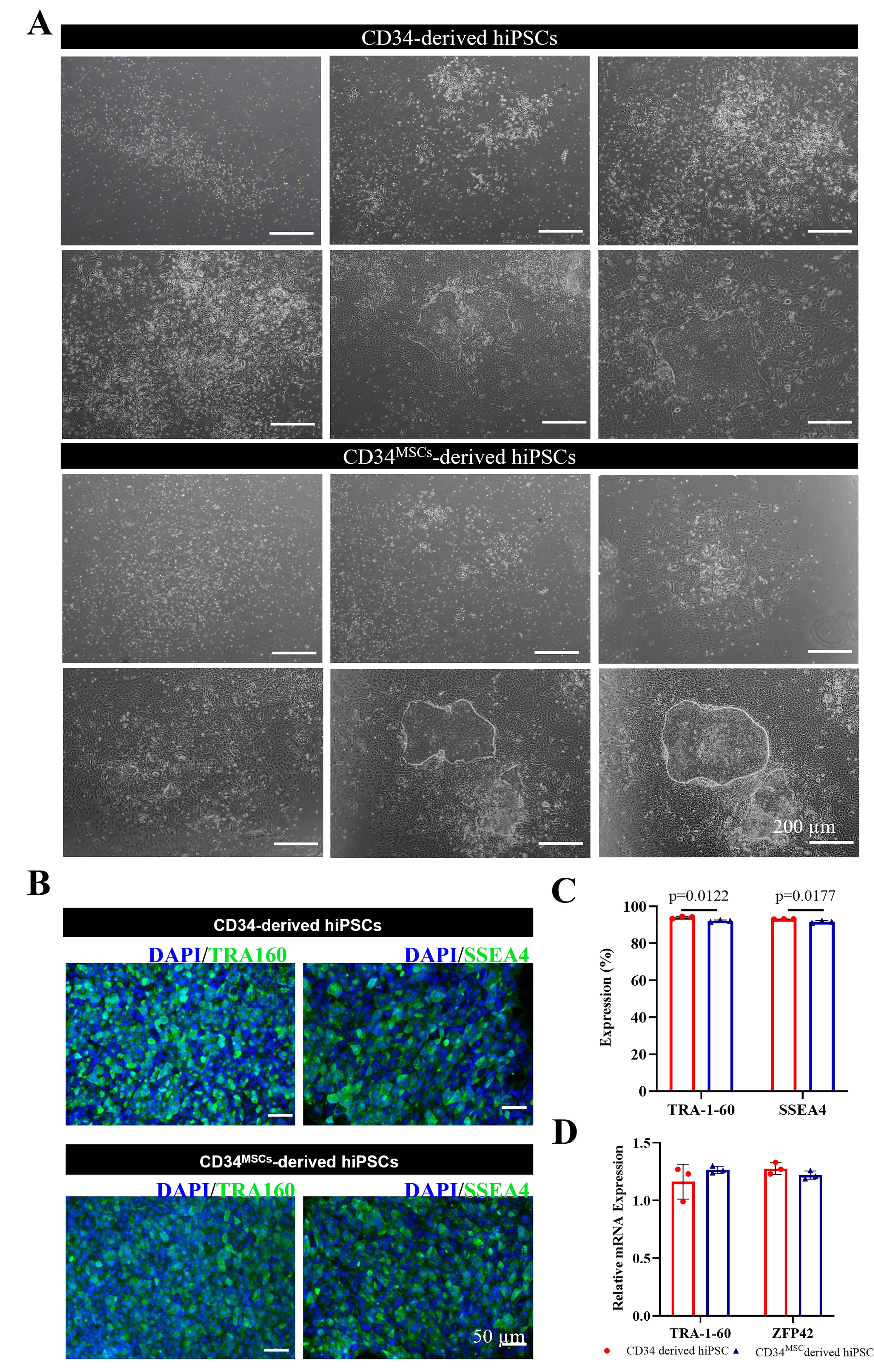
CD34MSC-derived hiPSCs show no difference in reprogramming efficiency and pluripotent marker expression.
Both hiPSC lines demonstrated pluripotency and were able to undergo differentiation into cells of the three germ layers
To confirm the pluripotency of the generated hiPSCs, the cells were induced to differentiate into cells of the three germ layers, namely, the cardiomyocytes (mesoderm), neuron (ectoderm), and early endodermal cells. Upon the induction of directed cardiomyocyte differentiation, spontaneously contracting cells were observed in 10 days (Supplementary Videos SV1, Video SV2). Immunocytochemistry staining confirmed the differentiated cells expressed cTnT and myosin light chain 2 ventricular, which were not different from the two groups of hiPSC lines (Fig. 4A). Similarly, dopaminergic neuron and early endodermal cell differentiation was performed on both groups of hiPSCs and the presence of mature dopaminergic neurons was confirmed by the expression of beta-III tubulin and TH on day 44 (Fig. 4B), and SOX17 and FOXA2 (Fig. 4C), with no difference in the differentiation ability despite the modifications with UC-MSC co-culture to the parental CD34+ cells.

Both hiPSC lines demonstrated pluripotency and lineage differentiation potential.
Transgene retention, karyotype stability, mycoplasma detection, and cell authentication of the generated hiPSCs
All the expanded hiPSCs at passage 18 showed normal karyotypes with 46,XX chromosomes (Supplementary Fig. S1, n = 3), regardless of the CD34 cell expansion method. Additionally, the hiPSCs were found to be transgene-free at 3–5 passages (Supplementary Fig. S2) and confirmed to be free from mycoplasma contamination (Supplementary Fig. S3). Cell authentication was performed using STR analysis to examine the loci D18S51, D21S11, TH01, D3S1358, Penta E, FGA, TPOX, D8S1179, vWA, CSF1PO, D16S539, D7S820, D13S317, D5S818, Penta D, and amelogenin, comparing the human-generated iPSCs with their parental CB unit CB023873, CB012604, and CB013104. The generated human iPSC lines in this experiment (control lines) were renamed to USMi001-A, USMi002A, and USMi003-A. The results revealed a complete loci match between the original CB samples and the generated hiPSCs at passage 18 (Table 1).
Short Tandem Repeats Analysis for USMi001-A, USMi002-A, and USMi003-A
The test samples profile showed 100% match with the following reference cell lines.
STR, short tandem repeat.
Discussion
Currently, the utilization of CB is predominantly restricted to the treatment of hematological disorders (Rafii et al., 2021). Given the growing preference for haploidentical transplantation over CB transplantation in clinical settings, the utilization of CB in clinical transplantation has been decreasing (Abberton et al., 2022; Iida et al., 2021; Niederwieser et al., 2022). To expand its therapeutic applications, CB can be repurposed to generate hiPSCs. However, low total nucleated cell count in CB can be a limiting factor to this effort.
The main issue with expanding HSCs outside the body is their high rate of asymmetrical division, which reduces long-term repopulating cells (Guenechea et al., 2001; Johnson et al., 2024). Since CB units already have few cells, further loss of CD34+ HSCs, reduced cell functions (Fietz et al., 1999; Hofmeister et al., 2007), and potential chromosomal changes (Ge et al., 2011) from expansion can make reprogramming and producing high-quality hiPSCs more challenging. MSCs help create the ideal environment for HSC growth, maintaining cell quality during expansion (Breems et al., 1998; Bucar et al., 2021; Wagner et al., 2007). This ensures that cells for reprogramming remain unaffected by in vitro changes that could harm hiPSC quality. In our study, MSC co-culture expanded CD34+ cells from revived CB by six times in 3 days, consistent with other studies (Corselli et al., 2013; Lau et al., 2017; Lin et al., 2020). Importantly, the hiPSCs we generated were functional, genetically intact, and matched the original CD34+ cells. Using synthetic matrices and growth factors instead of UC-MSC co-culture could simplify the process for clinical use.
Early hiPSC culture conditions are crucial for ensuring that fully reprogrammed, high-quality cells can grow and remain undifferentiated for expansion. We compared two commonly used matrices, Matrigel and Geltrex, both derived from mouse sarcoma cells. Although they are widely used for iPSC work, they contain animal-derived components, making them unsuitable for clinical applications under Good Manufacturing Practice standards. Xeno-free alternatives like laminin, vitronectin, retronectin, and 3D culture systems (Matsuo-Takasaki et al., 2023), have been proposed for clinical use.
Several combinations of reprogramming factors have been developed since 2006, when the Yamanaka factors—Oct3/4, Sox2, Klf4, and cMYC—were introduced (Takahashi and Yamanaka, 2006). Other groups have replaced Klf4 and cMYC with NANOG and Lin28 (Yu et al., 2007), while others used human telomerase reverse transcriptase (hTERT) and simian vacuolating virus 40 large T antigen (SV40 LT), or replaced Oct3/4 and Klf4 with T-cell leukemia/lymphoma protein 1A (Tcl-1a) (Masip et al., 2010). Many efforts focus on removing cMYC, a gene linked to cancer (Dang, 2012; Poli et al., 2018). While cMYC is dispensable for reprogramming (Wernig et al., 2008), it accelerates iPSC generation (Prieto et al., 2018; Rand et al., 2018). To lower cancer risk, L-MYC, with lower cancer-causing potential, has been used instead of cMYC (Nakagawa et al., 2010); or cMYC has been replaced by small molecules like valproic acid (Huangfu et al., 2008). In this study, we used the L-MYC strategy with a non-integrating episomal method to generate hiPSCs, a safe technique used in clinical trials (Bloor et al., 2020).
Interline variability has been shown to affect the characteristics of reprogrammed hiPSCs. This variability arises due to differences in reprogramming techniques (Kang et al., 2015), donor age (Lo Sardo et al., 2017), parental somatic cells (Kim et al., 2010), genetic diversity (Kyttala et al., 2016), and epigenetic variation (Nishizawa et al., 2016) of the donor cells. Notably, the propensity for differentiation is influenced more by donor-related variations than by the source of the cells (Terheyden-Keighley et al., 2024). A study suggests that approximately 50% of the variation in gene expression across the genome can be attributed to individual differences, primarily linked to chromatin regulators like Polycomb targets, which contribute to the variability in hiPSCs resulting from reprogramming (Carcamo-Orive et al., 2017). These factors affect hiPSC responses to small molecules and differentiation inducers among lines from different donors, thereby complicating efforts to standardize the manufacturing process, improve culture consistency, and enhance differentiation efficiency while reducing costs (Colter et al., 2021; Madrid et al., 2024).
The use of hiPSCs derived from CB is primarily focused on autologous applications. To expand their potential for allogeneic use, gene editing technologies like CRISPR/Cas9 can be employed to reduce immunogenicity. This may involve knocking down genes for major histocompatibility complex (MHC) molecules, which are key for antigen presentation and immune response (Sackett et al., 2016). Lowering MHC expression might help hiPSCs evade host immune recognition. Another approach is using immunosuppressive agents such as cyclosporine or tacrolimus, though these carry risks like increased infection or malignancy. Therefore, alternative strategies to promote immune tolerance and reduce reliance on immunosuppressive drugs are being explored. For effective therapeutic use, hiPSCs must be large in number, high-quality, and homogeneous, with stable genetic integrity. Evaluating hiPSCs from various sources is crucial to ensure consistency and quality, potentially requiring modifications to standardize these cells for clinical applications (Colter et al., 2021).
Our study also included STR analysis to compare the generated hiPSCs with the parental cells and demonstrated that our culture and processing retained the genetic identity (Sarafian et al., 2018) similar to the parental CD34 cells throughout, as analyzed at passage 18. Future study to extend STR analysis on differentiated cells will be particularly useful prior to clinical translation to avoid any unintended genetic drifts that may arise during the differentiation process (Keller et al., 2018). While we demonstrated trilineage differentiation across all generated hiPSCs, an in-depth assessment of efficiency and differences between the lines was not conducted. Mass testing of different iPSC lines for the applicability of designated differentiation protocols could aid in standardization, reducing interline variation and improving efficiency (Burridge et al., 2011; Thomas et al., 2022).
Additionally, routine karyotyping and cell quality assessment are necessary to ensure reproducibility and product consistency across different iPSC lines, which is crucial for realizing the potential of autologous therapy, precision disease modeling, and targeted medicine (Volpato and Webber, 2020). Spectral karyotyping (SKY) is a type of fluorescence in situ hybridization that uses multicolor chromosome painting to make each chromosome easy to identify and analyze. In hiPSCs, common abnormalities include extra copies (trisomy) of all or parts of chromosomes 1, 12, 17, 20, and X (Draper et al., 2004; Maitra et al., 2005; Martins-Taylor et al., 2011), or losses (monosomy) and structural issues like duplications, deletions, inversions, amplifications, and translocations (Mayshar et al., 2010; Olariu et al., 2010; Taapken et al., 2011). However, SKY has limitations in detecting small changes within the same chromosome and does not provide precise details on breakpoints or rearrangements. Giemsa (G)-banding, on the other hand, reveals staining patterns to detect chromosomal abnormalities and is a standard method for evaluating iPSCs (Wells et al., 2024). Additionally, single-nucleotide polymorphism analysis can detect copy number variations, unbalanced translocations, and mosaicism over 20% (Steventon‐Jones et al., 2022; Popp et al., 2018).
Conclusions
In conclusion, this study demonstrates that the expansion of CB CD34+ cells in UC-MSC co-culture prior to reprogramming does not compromise the generation or quality of hiPSCs. Further investigation is required to standardize the production process to ensure consistent and reproducible hiPSCs suitable for clinical therapy.
Authors’ Contributions
JTJJT, KLT and KYT conceptualized the experiment. FFR, YY, and MW conducted the experiments. FFR, GCO, NANMY and MNAAP performed the analysis. FFR and JJT wrote the article. SKC, MNAAR and JJT reviewed, revised, and finalized the article. All authors read and approved the article.
Funding Information
This project was funded by the USM-CryoCord Sdn Bhd Matching Grant (1001.CIPPT.8011102) and the Fundamental Research Grant Scheme (FRGS/1/2018/STG05/USM/03/3). JJT was also a recipient of the Dr. Ranjeet Bhagwan Singh Medical Research Grant 2022 and a research grant from ALPS Global Holding (304/CIPPT/6501080).
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.