
Abstract
Genome editing techniques have potential to revolutionize the field of life sciences. Several limitations associated with traditional gene editing techniques have been resolved with the development of prime editors that precisely edit the DNA without double-strand breaks (DSBs). To further improve the efficiency, several modified versions of prime editing (PE) system have been introduced. Bi-directional PE (Bi-PE), for example, uses two PE guide RNAs enabling broad and improved editing efficiency. It has the potential to alter, delete, integrate, and replace larger genome sequences and edit multiple bases at the same time. This review aims to discuss the typical gene editing methods that offer DSB-mediated repair mechanisms, followed by the latest advances in genome editing technologies with non-DSB-mediated repair. The review specifically focuses on Bi-PE being an efficient tool to edit the human genome. In addition, the review discusses the applications, limitations, and future perspectives of Bi-PE for gene editing.
Background and Historic Perspective of Genome Editing
Exogenous and endogenous stimuli can modify genetic information by altering nucleotide sequences (Matsumoto and Nomura, 2023). These alterations in the genes may be single-base pair (bp) modifications or multi-base pair rearrangements, deletions, and duplications (Eichler, 2019). All of these genetic changes may cause diseases in humans. Gene editing methods can improve our understanding of genetic diseases and lead to the development of new treatments. Back in the early days of classical genetics, when latest gene editing methods were not developed, scientists used traditional breeding techniques such as cross-breeding, selective breeding, and hybridization to introduce desirable traits into organisms. They employed genetic mapping to locate the genes responsible for the desired trait. This involved crossing different strains, observing the inheritance patterns of traits, and correlating them with specific chromosomal regions. Afterward, scientists increased the possibility of genetic modifications in laboratory animals by employing chemical and radiation mutagens. These techniques were expensive and time-consuming, even if they were useful. To gain a better understanding of the function of a gene, the field of molecular genetics employed reverse genetics. Reverse genetics is applied to introduce site-specific changes into genomic DNA by using DNA repair mechanisms.
Genome editing is the process of changing nucleotide sequences by deletion, addition, or substitution of particular bases into the genome. The concept of gene editing as a treatment of diseases or enhancing favorable genetic characteristics was conceptualized in 1953, when the double helix structure of DNA was proposed by Watson and Crick. Table 1 highlights the important discoveries made during the genome editing journey. The history of gene editing commenced in 1962 with the identification of the first restriction enzyme (i.e., HindII) in Haemophilus followed by its isolation and characterization in 1968. The restriction enzymes serve as molecular scissors by recognizing short nucleotide sequences in the DNA followed by breaking both strands of the DNA at their specific restriction site. During the early 1970s, these enzymes were used together with DNA polymerase and DNA ligase, to cut, alter, and study DNA in a predictable and reproducible way (Smith and Wilcox, 1970). The use of restriction enzymes is a remarkable landmark for the advancement of recombinant DNA technology. In 1972, Paul Berg used EcoRI to cleave DNA of SV40. The cleaved DNA was then introduced into the vector that was combined with chromosomal DNA fragments to form the recombinant DNA molecule. The use of restriction enzymes was later employed in other applications of gene editing methods. One such application is to produce delivery vehicles by cutting and ligating vectors with gene editing machinery such as transcription activator-like effector nucleases (TALENs) or cluster-regularly interspaced short palindromic repeats (CRISPR-Cas9), allowing effective delivery into specific cells.
Key Discoveries in the History of Genome Editing
CRISPR, cluster-regularly interspaced short palindromic repeat; TALEN, transcription activator-like effector nuclease; ZFN, zinc finger nuclease.
The development of site-specific recombinase technology in the 1980s was spurred by the discovery of the Cre-loxP system in bacteriophage P1. This innovative system leverages the Cre recombinase enzyme from bacteriophage P1 to precisely splice specific DNA sequences. Through its enzymatic activity, Cre facilitates site-specific DNA recombination, enabling the exchange of DNA segments between targeted sites within a DNA molecule. These sites, designated as loxP sequences, contain specific binding domains for the Cre enzyme, flanking a central region where genetic recombination occurs. This system is employed to make precise modifications such as targeted inversion, translocation, insertions, and deletions at specific sites within the genome (Sternberg and Hamilton, 1981).
Later, restriction enzymes were employed to develop double-strand break (DSB)-mediated and non-DSB-mediated gene editing methods. Zinc finger nucleases (ZFNs), TALENs, and CRISPR-Cas9 are examples of restriction endonucleases that are used in DSBs. Non-DSB-mediated methods include base editing and prime editing (PE). Genome editing has improved significantly since the introduction of ZFNs in the late 1980s, with the recent development of PE system in 2019 (Anzalone et al., 2019; Berg, 1990; Carlson et al., 2012; Jinek et al., 2012). Additional advancements in the original PE system (PE1), such as PE2-PE5 systems, PEmax, nuclease editors, next-generation PE system, and dual prime editors including the bidirectional PE (Bi-PE) system, have led to a paradigm shift in the field of life sciences, thereby creating opportunities for novel treatment interventions intended for genetic diseases (Anzalone et al., 2022; Anzalone et al., 2020; Matsumoto and Nomura, 2023). Scientists are persistently striving to improve the scope of genome editing as it holds exciting prospects for human genome modification with the aim to alleviate genetic diseases. This review discusses the conventional methods of gene editing that use DSB-mediated repair mechanisms and further explores the latest advancements in genome editing technologies that use non-DSB-mediated repair, such as base editing and PE (Fig. 1). The review focuses on Bi-PE, which involves the use of dual pegRNAs. It highlights the potential applications, limitations, and future directions of Bi-PE.
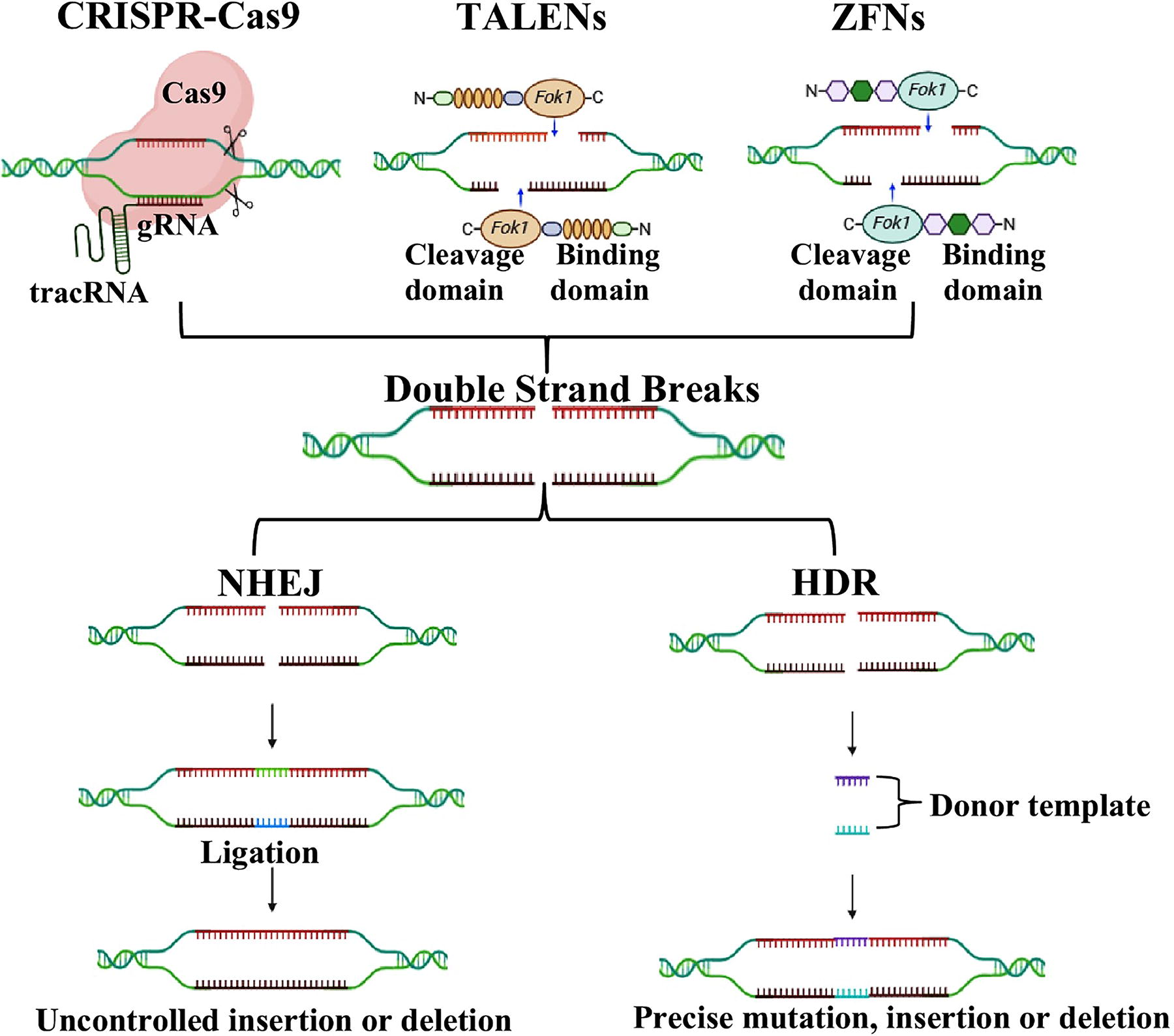
DSB-mediated gene editing. The CRISPR-Cas9 uses cas9 nuclease and guide RNA to cause double-strand breaks. TALENs use the FokI and TALE DNA binding domains as a tool to generate DSBs. ZFNs employ two FokI restriction enzymes and zinc-finger DNA binding domains to form DSBs. NHEJ and HDR fix the DSBs. DSBs, double-strand breaks; CRISPR, cluster-regularly interspaced short palindromic repeats; TALENs, transcription activator-like effector nucleases; TALE, transcription activator-like effectors; ZFNs, zinc finger nucleases; NHEJ, non-homologous end joining; HDR, homology-directed repair.
DSB-Mediated Gene Editing
The first steps in homologous recombination (HR)-based precision genome editing involved inserting an external DNA template into desired cells. This technique has been successfully implemented for creating knock-in cell lines and genetically modified mice. Nevertheless, the rate of HR between donor DNA and target DNA is very low when there is no DSB. This limitation has prevented the use of this approach in clinical applications. However, when a DSB and a donor DNA template are both positioned at the exact location in the genome where the edit is desired, the efficiency of HR can be significantly improved. This method depends on precise breaking of the double-strand DNA of the target gene. ZFNs, TALENs, and CRISPR-Cas9 are the three most widely used site-specific techniques that edit the genome by employing DSBs (Zheng et al., 2024). The comparison of gene editing techniques has been described in Table 2.
Comparison of Gene Editing Techniques
Bi-PE, bidirectional prime editing; DSB, double-strand break; PAM, protospacer-adjacent motif.
Zinc finger nucleases
ZFNs marked the beginning of DSB-mediated genome editing technology. ZFNs have the ability to choose and accurately target any gene in an organism. Zinc fingers work as the transcription factors, where each finger selectively identifies a sequence of 3–4 DNA bases. ZFN is composed of a DNA-cleaving domain that cuts DNA at the required position and a zinc finger DNA-binding domain that recognizes a specific DNA site (Bibikova et al., 2001). The DNA-cleaving and DNA-binding domains of ZFN combine to provide a specific and highly accurate set of genomic scissors. These domains interact with DNA to generate DSBs at specific DNA loci. DSBs are typically repaired by the cellular mechanisms, namely, HDR (homology-directed repair) and NHEJ (non-homologous end joining). High-fidelity HDR is a specialized mechanism for repairing DNA that relies on a homologous template to accurately guide the repair process. In contrast, NHEJ is a repair mechanism where DSBs are joined together without the need of homologous sequence. Since their first discovery, ZFNs have gathered significant interest and demonstrated great potential for genome editing. (Carroll, 2011; Carroll, 2014). However, the limitations of the ZFNs system include low efficiency, nonspecific binding, off-target cleavage of DNA sequences, targeting only a single site at a time, high cost, and time-consuming manufacturing. Many nonspecific cleavages can damage cells by causing unwanted mutations and off-target effects (Khalil, 2020).
Transcription activator-like effector nucleases
TALEs were first discovered in 2009. TALEs are endogenously produced proteins within the Xanthomonas bacteria where they regulate gene transcription in plant host cells. Scientists later developed the TALENs by fusing the TALE domain with Fok1 restriction endonuclease for a more precise edit (Boch and Bonas, 2010). TALENs are made up of a FokI nuclease domain and a customized DNA-binding domain as compared with ZFNs (Christian et al., 2012). The customized DNA-binding domain is made of a variable number of conserved repeats, normally between 33 and 35, which selectively identify certain nucleotides (Bhushan et al., 2016). With the highest specificity of target sequence cleavage compared with ZFNs, it is now possible to target any DNA sequence inside the genome of an organism. Compared with ZFNs, the TALENs method shows greater efficiency in generating DSBs in both pluripotent stem cells and somatic cells (Cong et al., 2013). TALENs show lower toxicity in human cell lines because of their fewer off-target effects, specificity, transient expression, and improved design (Huo et al., 2019). Phage-assisted continuous evolution (PACE) has been implemented to increase the specificity of TALENs using TALEN-like directed evolution of DNA-binding proteins. Using DNA-binding PACE is an extremely versatile approach to develop tailored strategies for DNA-binding domain, thereby increasing the accuracy of the genome editing tools (Huo et al., 2019; Khan, 2019). Despite these advantages, TALENs encountered some limitations including their larger size, sensitivity to target DNA methylation, require the production of unique proteins for each specific objective, low efficiency, and limited flexibility to target sequence (Huo et al., 2019).
CRISPR-Cas9 system
The groundbreaking utilization of the Cas9 protein for RNA-guided DNA cleavage was first reported in 2012 (Jinek et al., 2012). A pivotal discovery in this study revealed that Streptococcus pyogenes-derived Cas9 can efficiently cleave DNA in a controlled laboratory setting using trans-activating RNA (tracrRNA) and CRISPR RNA (crRNA) complementary to the target DNA (Anzalone et al., 2020). The Cas9 enzyme features two distinct nuclease domains: NHN and RuvC. RuvC targets the noncomplementary strand whereas HNH targets the complementary strand, hence simultaneously cleaving the DNA duplex. Specifically, altering a nuclease domain converts Cas9 into a nickase, sometimes known as nCas9, which causes single-strand breaks or “nicks” in DNA. Single strand breaks creatded by nCas9 result in more exact editing and less off-target effects, as in base editing and PE. The Cas9 enzyme starts R-loop development using a protospacer-adjacent motif (PAM), then cleaves DNA. PAM is a short 2–6 nucleotide sequence within the target DNA that varies between species and Cas enzymes, such as NGG in Streptococcus pyogenes (SpCas9) and NGA in Staphylococcus aureus (SaCas9). Moreover, tracrRNA and crRNA can be merged into one guide RNA (sgRNA), therefore guiding the Cas9 protein to particular DNA regions (Anzalone et al., 2020).
The CRISPR-Cas9 system was initially identified in bacterial cells as a defense mechanism against foreign infections. Further research revealed CRISPR sequences in 20 diverse organisms, leading to the discovery of various CRISPR-Cas systems. These systems evolved due to differences in CRISPR locus design and the presence of diverse Cas genes. The latest classification categorizes CRISPR-Cas systems into two distinct classes. Class 1 encompasses Type I, Type III, and Type IV systems, which rely on multiprotein complexes and CRISPR RNA (crRNA) to perform their functions. In contrast, Class 2 comprises Type II, Type V, and Type VI systems, characterized by a single multidomain protein. The Type II CRISPR-Cas9 system has been extensively investigated and used in genome editing. Its simple design has made it a desirable tool for scientists and researchers trying to precisely and accurately alter genomes (Arif et al., 2023).
The CRISPR-Cas9 system comprises two essential components: guide RNA (gRNA) and CRISPR-associated (Cas) protein. This genome editing technique involves a three-stage process: recognition, cleavage, and repair. gRNA is a 17–20 nucleotide sequence specifically designed to target a unique region within a gene. This sequence is complementary to the target DNA sequence, adjacent to a PAM, ensuring precise targeting. PAM is often “NGG,” for recognition by SpCas9 (Streptococcus pyogenes Cas9), a commonly used CRISPR-Cas system. However, other Cas proteins recognize alternate sequences such as TGG, AGG, GGG, and CGG, which serve the similar purpose. The presence of a PAM, typically 3–6 bps downstream of the Cas cleavage site, is important for the interaction between Cas and its target. After the gRNA-Cas9 complex forms, the gRNA guides the Cas enzyme to target site where Cas induces a DSB in the DNA molecule, particularly three bps upstream of the PAM sequence. Following the DSB, the cell’s natural repair machinery is activated, and the break can be repaired through two main mechanisms, that is, HDR and NHEJ (as in ZFNs and TALENs) (Asmamaw and Zawdie, 2021). The NHEJ cellular repair process is the dominant and remarkably effective method as it has the highest level of activity in cells. However, errors may occur, resulting in small and random insertions or deletions (indels) in the area of the cleavage site. This may lead to the occurrence of premature stop codons or frame-shift mutations. HDR necessitates the utilization of a DNA template that is precisely or substantially identical from the target sequence, making it an exceedingly precise method (Liu et al., 2018; Yang et al., 2020). The CRISPR-Cas system is considered to be easy to use, target-specific, multipotential, and more accurate than ZFNs and TALENs (Choudhery et al., 2024a; Choudhery et al., 2024b). However, this system also has several limitations including off-target effect, unavailability of PAM in the desired gene, introduction of DSBs, and lack of potential to edit specific loci. These challenges need to be addressed for the development of optimized CRISPR-Cas systems (Arif et al., 2023). (Gaudelli et al., 2017).
Non-DSB-Mediated Genome Editing
Most genome editing approaches produce DSBs that results in random changes, which can include insertions, deletions, and translocations at the targeted region in the genome. Gene correction is generally achieved with additional external DNA repair templates, coupled with the use of the mechanisms of HR, which have low efficiencies. The alternative approach is non-double strand-mediated genome editing (non-DSB), which can possibly make edits to the desired locus without the need for DSBs. In this section, we covered the methods of base editing and PE, as well as its advances, with a specific focus on Bi-PE. It is pertinent to note that recent research indicates the possibility of DSBs with cytosine base editors (CBEs) (Huang et al., 2024). This has been discussed further below.
Base editing
The original CRISPR-Cas9 system creates dsDNA breaks for desired mutations at DNA loci, which was hampered by NHEJ and HDR, creating undesirable indels. To bypass the above-mentioned limitation discussed in the above section, CRISPR-Cas9 was modified to develop base editors (BEs). BE is a powerful genome-engineering tool, leveraging CRISPR technology to induce precise single-nucleotide modifications (Gaudelli et al., 2017; Komor et al., 2016). Adenine base editors (ABEs) and CBEs are the two principal types of BEs. The ABEs have the ability to convert adenine (A) into guanine (G) and thymine (T) into cytosine (C). Conversely, CBEs have the ability to induce mutations by converting guanine (G) to adenine (A) and cytosine (C) to thymine (T). BEs enable precise introduction of the four transitional mutations, C → T, T → C, G → A, and A → G to the genome (Arif et al., 2023).
This innovative approach, first introduced in 2016, exploits Cas9’s targeting ability to recruit another DNA-altering enzyme to specific genomic locations (Komor et al., 2016). Cytidine deaminase, more especially rAPOBEC1 (apurinic/apyrimidinic endonuclease 1), catalyzes deamination in the base editing process. This enzyme converts exocyclic amine found in cytosine to uracil. DNA polymerases may incorrectly read deaminated cytosine as thymine during DNA repair and replication since uracil and thymine show similar hydrogen bonding patterns. CBEs enable specific, programmable C•G to T•A alterations without generating double-strand DNA breaks by combining Cas9’s targeting accuracy with APOBEC enzymatic activity. The preference of the APOBEC enzyme for single-strand DNA enables correct editing. By selectively targeting the single-strand DNA of the Cas9 R loop, APOBEC1 avoids deaminating cytosines that are adjacent to the target base or present elsewhere in the genome, thereby minimizing off-target effects. Komor et al. (2016) developed the first BE, that is, BE1, by combining a catalytically inactive Cas9 (dCas9) with rAPOBEC1. While BE1 demonstrated effective cytosine deamination in vitro, its editing efficacy in the HEK293T cell culture was below 10%. In order to enhance the efficacy of cellular editing, the authors implemented two modifications to promote the repair of cellular DNA. BE2 was created by adding a uracil glycosylase inhibitor (UGI) peptide to the editor. This peptide inhibits cellular UDG (uracil-DNA glycosylase). UDG catalyzes uracil removal from DNA by hydrolyzing its N-glycosidic bond of the base, initiating the BER pathway. This DNA repair process restored the target base to its original cytosine form. BE2 caused an increase in C•G-to-T•A mutations through repression of UDG and interference with the conversion of uracil back to cytosine. Finally, Komor et al. (2016) enhanced the functionality of the Cas9 HNH nuclease domain by restoring its catalytic activity, resulting in the creation of BE3. The Cas9(D10A) nickase of the BE3 editor selectively cleaves the unprocessed DNA strand, while the edited strand remains unaltered. If an unedited strand contains a nick, it will be repaired by utilizing the modified strand as a template. This results in the permanent incorporation of a C•G to T•A mutation onto both strands of DNA (Komor et al., 2016). The discovery of the first CBEs prompted subsequent development of ABEs in 2017 (Gaudelli et al., 2017). In ABEs, adenine within the editing window is deaminated to produce inosine (I), which is subsequently recognized as guanine by DNA repair machinery, resulting in an A-to-G substitution after DNA replication and repair. The ABE mechanism yields little A-to-C, T, or indel byproducts because glycosylase activity on the “I” base in eukaryotic cells is reduced or insufficient. When cells are exposed to exogenous or endogenous substances, they experience nitrosative stress and natural hydrolysis, which causes the deamination of A to I and I:T mismatches, activating both alternative and base excision repair mechanisms (Molla et al., 2020; Zeng et al., 2022).
Activation-induced cytidine deaminase (AID) was used by other BEs to change C into the other three bases in a 4-bp or 100-bp catalytic frame. Somatic hypermutation and class-switch recombination are two mechanisms that AID stimulates in activated B cells to generate Immunoglobulins (Igs) with high affinity. Ig genes undergo AID deammination from C to U, which UDG excises to form apurinic/apyrimidinic (AP) sites. Rev1 and other translesion synthesis (TLS) polymerases process these sites to enhance Somatic hypermutation (SHM). C is uniquely located opposite U and AP sites in Rev1, a Y-family TLS polymerase. Instead of A, G, or T, Rev1 substitutes C over AP damages, in contrast to typical replicative DNA polymerases. In the S phase, but not in G1, Rev1 is recruited to a basic site produced by UDG and excises uracils formed by AID. When replication forks stop, Rev1 adds Cs to get around AP sites. AID-UDG might be attached to the N-terminus of a dCas9 with Rev1 at the C-terminus in order to guide it to a certain C locus. One major problem with this process is that Rev1 cannot attach and use the AP site-containing strand as its inverse. Rev1 cannot attach to the template strand following dCas9 release because the single-strand bubble closes, independent of whether nCas9 produced a nick in the opposite strand (Gajula, 2019).
Interestingly, custom genomic locus targeting was achieved by coupling Escherichia coli DNA polymerase I (PolI5M) to the C terminus of nCas9 and employing suitable sgRNAs (Halperin et al., 2018). In EvolvR, a nick is created at the protospacer after nCas9 binds to the target locus. The error-prone synthesis of the strand opens the nick and introduces sequence variation within 17 nucleotides as PolI5M is released. It is possible to use the EvolvR system to diversify the nucleotides of these novel domains, and then to use PACE to increase the number of PAM recognition sites in Cas9. Through multiple rounds of optimization, this method has the potential to produce the Rev1 variation that is most compatible with any target locus for C→G editing, while also striking the ideal balance between C integration across AP sites. Important improvements to this method include the use of mutant C deaminases that may either function on a tiny window of one to two nucleotides or selectively deaminate a C when many Cs are present in the editing window, thereby avoiding bystander base editing (Gajula, 2019).
Base editing is an important step for the understanding of human pathophysiology as it enables precise deletions, insertions, or substitutions of nucleoties causing genetic diseases. However, the risk of inadequate editing and off-target effects make their use limited. In addition, potential to generate the eight required transversion mutations (T → A, A → T, T → G, C → A, G → C, A → C, G → T, C → G) is challenging (Arif et al., 2023). Though earlier research indicated that CBEs do not cause DSBs, new experimental data showed the opposite outcomes. The results of this work show that CGBEs, or C-to-G BEs, produce intermediate DSBs. To minimize DSB-triggered events without compromising base substitutions, it is worth mentioning that protecting target sites with the suicide enzyme HMCES (5-hydroxymethylcytosine binding, embryonic stem-cell-specific) reduced CGBE-initiated DSBs. The suicide enzyme HMCES protects the AP sites produced by deaminase. The suicide capability, which is acquired by CGBEs combined with HMCES, could add an extra safety. However, the suicide enzyme does not entirely remove DSBs. More research is required to optimize CGBEs in the future, which could lead to the development of an ideal transversion editor with fewer background byproducts and more transversion purity (Huang et al., 2024).
PE system
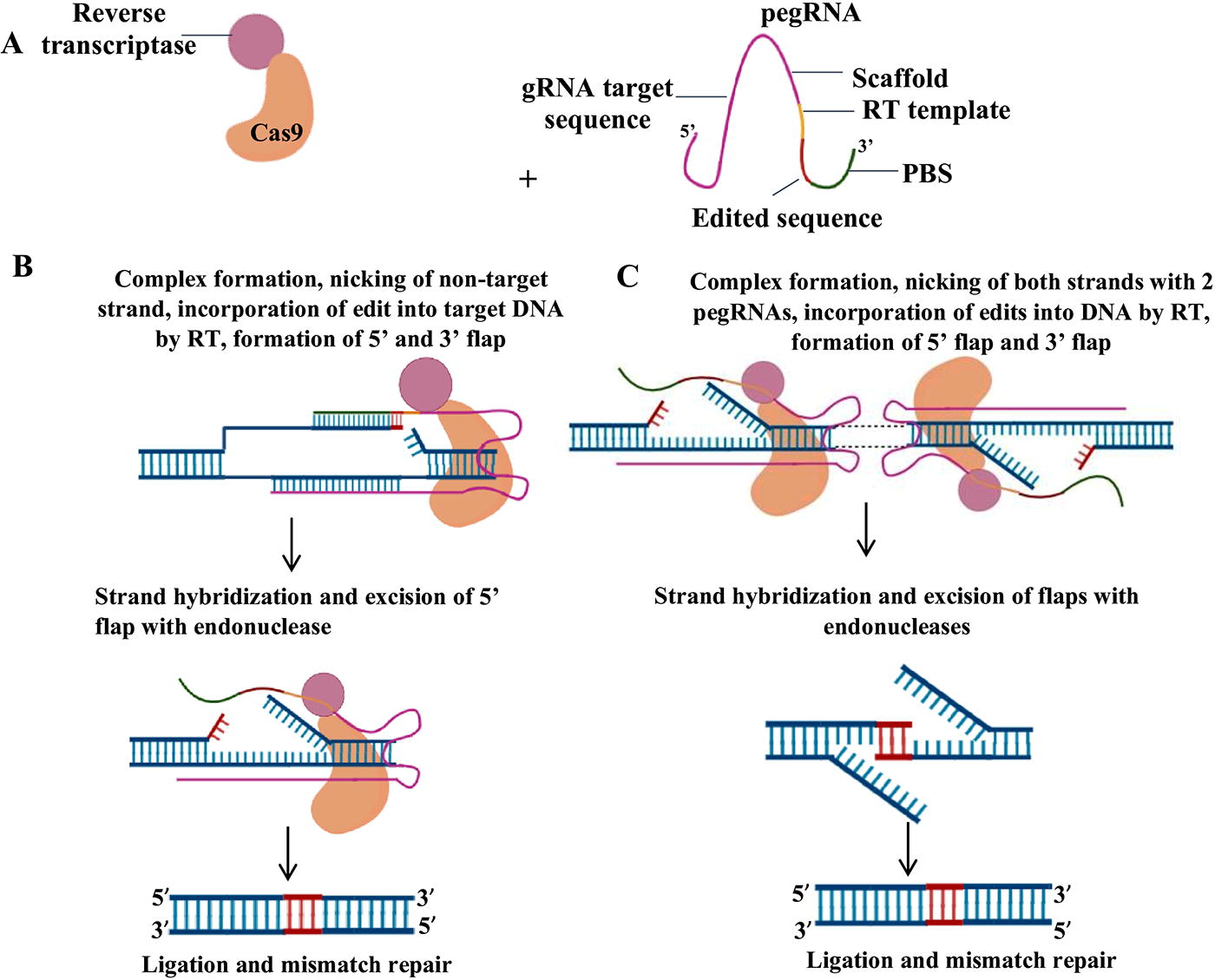
PE was developed at the laboratory of David R. Liu at the Broad Institute in 2019. The PE system broadened the scope of DSB-independent gene editing by enabling all 12 base conversions (i.e., T→C, A→G, C→T, G→A, T→A, A→T, C→A, G→C, T→G, A→C, C→G, and G→T), as well as other specific insertion and deletions of gene part, without requiring DSBs or donor DNA. PE has the potential to correct up to 89% of genetic disorders if employed correctly (Anzalone et al., 2019). The PE technique is a very adaptable and potent tool for manipulating genes with great accuracy, which has gained significant interest in its further development. The development process of the PE system led to many enhancements in its components and mechanisms. Figure 2 describes various components of prime editors as well as the traditional PE mechanism. Various classes of prime editors, such as PE1, PE2, PE3, PE4, PE5, nuclear prime editors, PE2-PE5 max system, next-generation prime editors (PE6, PE7), engineered pegRNAs (epegRNAs), and bidirectional prime editors (Bi-PEs), were created to precisely edit the genome. The key differences between original PE and Bi-PE editing have been given in Table 3.
Key Differences Between Original Prime Editing and Bidirectional Prime Editing
M-MLV, Moloney murine leukemia virus; RT, reverse transcriptase.
Initial modifications in the PE system
The first PE system consists simply of a pegRNA and the prime editor. The pegRNA includes 5′ gRNA and 3′ reverse transcriptase (RT) template with primer binding site (PBS) and scaffold in-between. The prime editors include a catalytically impaired nickase (Cas9 H840A) combined with the wild-type Moloney murine leukemia virus (MMLV) RT enzyme. The Cas9 nickase was created by replacing histidine with alanine at position 840. H840A mutation makes RuvC domain of Cas9 inactive while HNH domain retains its activity. The prime editor, guided by pegRNA, forms a complex with the targeted genome. This complex opens the DNA double helix, exposing the nontarget strand as single-strand DNA. Cas9 introduces the nick 3 bp upstream of the PAM site, resulting in the formation of a 3′ flap. The PBS of the pegRNA interacts with the 3′ flap of target DNA and forms a DNA/RNA hybrid. This site of DNA, that is, 3′ end serves as a primer for the synthesis of a new DNA strand. The RT uses RT template part of pegRNA as template to reverse transcribe it in the DNA strand, thereby extending the 3′ flap in 5′ to 3′ direction. The extension sequence, that is, the RT template contains the required edits, which are copied into original strand during reverse transcription. Cellular flap structure-specific endonucleases such as flap endonuclease 1 (FEN1) removes the unedited 5′ flap of the original strand due to its inherent preference of breaking the phosphodiester bond. Ligase 1 then ligates the edited 3′ flap onto the original phosphate backbone. A second guide RNA (gRNA) nicks the opposite (target) strand at adjacent positions to regulate the cellular replacement of the unedited strand (original). The cellular repair machinery replaces the original strand with the newly edited strand. The edited strand is preferentially used as the template for repair, increasing the efficiency and accuracy of the PE process (Arif et al., 2023).
A new system called prime editor 2 was created when PE1 was manipulated to contain the pentamutant RT enzyme. In contrast to PE1, the PE2 method was more systematic in achieving specific deletions and insertions. It demonstrated a 5.1-fold increase in editing efficiency (Anzalone et al., 2019). Despite this improvement, the edits made by PE2 still needed to be corrected because the cell’s natural DNA repair mechanism can correct the edited strand, potentially removing the intended changes. This issue was addressed by developing the PE3 system. PE3 introduced an extra single guide RNA (sgRNA) during DNA heteroduplex resolution. The purpose of this sgRNA is to mimic the pegRNA-induced altered sequence rather than the original allele. It guides the fusion proteins Cas9 nickase to nick the unedited strand opposite the initial nick. Nicking the nonedited strand permanently installs the edit because the normal cellular repair mechanism copies the information in the edited strand from the complementary strand. The nicking of the unedited strand, on the contrary, might result in the formation of extra undesirable indels, which is one of the disadvantages of this approach. Scientists have reduced this issue by developing the PE3b, which has the maximum editing efficiency when compared with PE3. In the PE3 method, the second nick is introduced 14–116 nt from the original pegRNA nick site in the unedited strand. The PE3b method limits the formation of extra undesirable indels by allowing the nCas9-sgRNA complex to match the edited sequence only and induced a nick on the nonedited strand. Prime editor 4 makes use of the same machinery as PE2, but it additionally incorporates a plasmid that encodes for mutL homolog 1 (MLH1), a dominant negative mismatch repair (MMR) protein. The dominant negative MLH1 inhibits endogenous MLH1, which reduces the cellular MMR response and increases the PE efficiency. Prime editor 5 uses PE3′s machinery and a dominant negative MLH1 plasmid. Similar to PE4, this enhances the efficiency of PE by inhibiting the endogenous MMR response. The transient expression of a modified protein that inhibits MMR improves the efficiency of substitution, insertion, and deletion by an average of 7.7 times compared with the PE2 system, and 2.0 times compared with the PE3 system. However, inhibiting the MMR protein can increase the indels ratio. The PE nuclease (PEn) utilizes a fully active Cas9 enzyme, which cuts both DNA strands, unlike traditional prime editors, which use a less active nickase enzyme that only cuts one strand. The PEn enzyme can successfully edit sites that are difficult or impossible for traditional prime editors to modify, but it also tends to introduce more unintended changes or off-target effects because the fully active Cas9 enzyme can make mistakes and edit nontargeted parts of the genome (Anzalone et al., 2019; Arif et al., 2023). Research suggests that installation of silent mutations near the intended edit might improve PE outcomes by reducing MMR (Chen et al., 2021).
Development of epegRNAs
Optimizing pegRNAs for PE presents a major challenge. Constructing hundreds of pegRNAs for a single desired change usually requires significant experimentation to find the most successful variant (Zeng et al., 2024). The 3′-extension of the pegRNA, which contains the RT template and PBS, is crucial to the pegRNA design and the success of PE. However, cellular ribonucleases can degrade this 3′-extension, reducing primary editing efficiency. Researchers added structured RNA motifs from viral pseudoknots such as evopreQ1 ormpknot into the 3′-end of pegRNAs to address this issue. This change increased pegRNA stability and protected 3′-extension from degradation. In primary human fibroblasts and human cell lines without increasing off-target effects, the tailored pegRNA, known as epegRNA, improved PE efficiency by three–four times (Nelson et al., 2022).
By introducing different motifs into the 3′-extended region, researchers have made important progress toward improving pegRNA stability. Adding stem-loop aptamer, viral exoribonuclease-resistant RNA motifs, guanine quadruplex (G-quadruplex) structures, or human telomerase RNA sequences has shown in recent research to enhance pegRNA performance. These changes have produced efficiency increases similar to those obtained with epegRNA, therefore extending the opportunities for optimal PE in mammalian cells (Feng et al., 2023; Li et al., 2022a; Zhang et al., 2022). Furthermore, Li et al. enhanced the stability of the pegRNA secondary structure by substituting each non-C/G pair with a G/C pair in the second stem loop of the pegRNA scaffold. This modification resulted in a 2.77-fold improvement in the efficiency of insertions and deletions (Li et al., 2022b). Interestingly, researchers have also developed a split pegRNA system in which a traditional sgRNA guides the nSpCas9 enzyme and a separate linear or circular PE template RNA, which includes the PBS and RTT, directs the RT to the desired location through the binding of MS2 coat protein and MS2 aptamer. The split pegRNA method demonstrates a comparable efficiency to the standard pegRNA, in which the spacer and template sequences combine in a single pegRNA (Liu et al., 2022). The efficiency of PE can be significantly enhanced by using epegRNAs. A common problem identified with conventional pegRNAs is the degradation of the 3′ end, resulting in a reduction of PE efficiency. epegRNAs are designed to prevent degradation by including a structured RNA motif at their 3′ end.
PEmax systems with optimized prime editors
The PE2max, PE3max, PE4max, and PE5max are the modified versions of PE2, PE3, PE4, and PE5, respectively. The PEmax design presents four improvements in the original editor, including use of codon, refinement of the nuclear localization signals (NLSs), and linkers, and incorporation of two Cas9 mutations previously found to enhance Cas9 nuclease activity, namely, SpCas9 R221K and N394K mutations. The specific set of codons commonly used in E. coli genes encoding amino acid sequences is comprised of the codons used in the PEmax system. The PEmax system showed a 2.5-fold increase in the average frequency of desired editing in HeLa cells and a 1.2-fold increase in HEK293T cells. This increase was observed at seven distinct loci compared with the PE2 editor design. Average edit/indel purity for PE3max and PE5max was slightly lower, approximately 1.2-fold lower relative to those of PE3 and PE5, respectively. The increased nickase activity resulting from the SpCas9 point mutations in the PEmax design could be responsible for this modest decrease (Chen et al., 2021).
Researchers have further enhanced the PE efficiency by combining the PE4max and PE5max systems with epegRNAs, which include an extra 30 RNA structural motif. epegRNAs increased PE4max editing efficiency with an average increase of 1.5-fold in HEK293T cells and 2.5-fold in HeLa cells, while maintaining the purity of the edits and indels. Similarly, epegRNAs increased PE5max editing by 1.4 times compared with standard pegRNAs in HeLa cells and by 1.1 times in HEK293T cells, compared with standard pegRNAs (Nelson et al., 2022). The integration of all the improvements to PE methods (MLH1dn, PEmax, and epegRNAs) has the potential to significantly increase the outcomes of PE. The PEmax method also has drawbacks, such as the complex synthesis of pegRNA, which can be expensive and time-consuming. PEmax, like other gene editing approaches, can produce off-target effects. While knock out of MMR activity enhances PE, the resolution of a heteroduplex intermediate without MMR remains unknown. Although PEmax allows for larger modifications than previous PE systems, deletions and insertions are still limited in size (Chen et al., 2021).
Next-generation prime editors
Next-generation prime editors include the P3 and PE7 systems. PE6 systems were developed with the PACE circuit for prime editors (PE-PACE). PACE is used to develop Cas9 nickase and compact RT domains that enhanced the PE efficiency. The PE6 system has evolved into nine subtypes (PE6a–PE6i). Escherichia coli Ec48 retron RT was evolved to produce PE6a, and Schizosaccharomyces pombe Tf1 retrotransposon RT was developed to produce PE6b, using PE-PACE (Doman et al., 2023). Building on previous iterations, researchers engineered PE6c by combining mutations from rdTf1 with the Tf1 RT found in PE6b. Similarly, PE6d emerged from integrating key mutations from PE2 with MMLV-RT ΔRNase H. Further innovations led to the creation of PE6e-g, which combines optimized SpCas9 variations with MMLV-RT ΔRNase H (Doman et al., 2023). Additional improvements to the Cas9 nickase domain yielded PE6f-i (Chen et al., 2021).
PE6 variants offer compact designs without compromising efficiency, comparable to their PE1–PE5 predecessors. These variants present unique advantages, particularly in specific applications. For instance, the smallest variant, PE6a, is ideal for use cases where delivery size is a limiting factor (Doman et al., 2023). PE6b enhances PE efficiency when MMLV-RT-derived prime editors are ineffective, while PE6c and PE6d excel with complex reverse transcription templates. Furthermore, PE6e-g boosts efficiency at specific sites. Advancement of PE6f-i has improved PE efficiency by up to 1.8-fold (Chen et al., 2021). The improved RT and Cas9 nickase domains of PE6 variants likely can be synergistically combined with each other as well as with MMR evasion strategies, epegRNAs, and the PEmax architecture. This combination results in cumulative advantages, making these primary editing systems the most advanced in the field. The PE tools demonstrate outstanding efficiency in therapeutically significant cell types such as patient-derived fibroblasts primary human T cells (Chen et al., 2021; Nelson et al., 2022).
Recently, scientists have developed the PE7 prime editors through enhancing PE with an endogenous small RNA-binding protein. Scientists have determined that La is an essential cellular determinant to enhance the efficiency of PE in different systems such as PE2, PE3, PE4, and PE5, and types of edits such as substitutions, insertions, and deletions. La binds to polyuridine tracts found at the 3′ ends of RNA molecules, thereby protecting them from degradation. By fusing the N-terminal domain of La to a PE protein, termed PE7, the scientists have improved PE efficiency significantly. This editor demonstrated enhanced performance with both expressed pegRNAs and engineered pegRNAs as well as synthetic pegRNAs optimized for La binding. Evaluation of the improved PE7 in disease-relevant cell types further showed substantial improvements in editing efficiency in several genomic targets linked to conditions such as sickle cell disease and HIV infection (Yan et al., 2024).
Dual prime editors
Dual-pegRNA systems were developed to improve the PE efficiency by enabling the editing of large DNA sequences, even gene-sized (Anzalone et al., 2022). These dual-pegRNA systems include dual-pegRNA, twinPE, PRIME-Del, homologous 3′-extension-mediated prime editor (HOPE), glycosylase-reverse transcriptase activation-deaminase-nucleotide (GRAND), and Bi-PEs (Choi et al., 2022; Jiang et al., 2022; Lin et al., 2021). These systems differ in the alteration of the original DNA sequence between the two breaks and position of complementarity between the two newly synthesized DNA strands. In twinPE and GRAND, the intra-nucleotide sequence is eliminated and substituted with the novel sequence encoded by pegRNAs. The complementary strands generated during reverse transcription can spontaneously anneal. PRIME-Del and Bi-PE have similarities with twinPE, with the distinction that the newly created DNA strands not only complement each other but also complement the sequence located upstream of the nick on the opposite DNA strand. Unlike the other approaches, the HOPE does not remove any sequence between the two nicks. Dual-pegRNAs improved both efficiency and accuracy of the PE system by targeting both strands of DNA. Dual-pegRNAs provide flexible genome editing, encompassing the ability to make both small or large deletions and insertion of large fragments (Choi et al., 2022; Jiang et al., 2022; Lin et al., 2021). Current review focused on the development, therapeutic potential, limitations, and future directions of Bi-PE.
Bi-PE for Efficient Genome Editing
Anzalone and colleagues introduced the Bi-PE system in 2021 (Adikusuma et al., 2021). Bi-PE increased the efficiency of PE by combining two pegRNAs, each of which selectively targets a distinct DNA strand to generate the desired genetic alteration. The paired pegRNAs function by generating complementary 3′ flaps that can bind together to form an intermediate structure. This intermediate structure consists of 5′ overhang from the original DNA sequence and 3′ overhang from the new DNA sequence that is joined together. The new sequence is replaced by the complementary 3′ flap sequences following the removal of the later and the joining of the breaks (Fig. 2). The method may substitute, insert, or delete a DNA sequence by modifying the template. The accuracy and efficiency of replacing and deleting large sequences are higher in comparison with previously described PE systems. Nevertheless, Bi-PE encounters difficulties when it comes to DNA insertions that exceed 100 bps in length. Remarkably, the integration of serine recombinase, nuclease editors, and epegRNAs with the Bi-PE system facilitated the targeted inversion and incorporation of large DNA sequences and enhanced the overall editing efficiency (Anzalone et al., 2022).
Integration of site-specific recombinases with Bi-PE system
Serine recombinase is an enzyme that stimulate the targeted incorporation of mobile genetic material into bacterial genomes by site-specific integration. This enzyme is able to precisely locate, attach, and cleave specific sites in DNA and, upon rearranging, the regions before religating the strands. Bi-PE can include recombinase recognition sequences. Upon inclusion of such sequences, recombinases may facilitate large-scale modifications (Yarnall et al., 2023). Anzalone and his colleagues have discovered that applying a combination of recombinases and Bi-PE is a very promising tool to enhance the insertion capacity of Bi-PE. The ability of recombinase-guided Bi-PE to create genetic modifications can also be applied for horizontal gene transfer with a purpose of improving single-gene characteristics (Anzalone et al., 2022).
Nuclease-based Bi-PE system
Using nSpCas9 (H840A), traditional nickase-based PE techniques need complex navigation through DNA repair processes, hence increasing the probability of either unsuccessful or unwanted editing (Anzalone et al., 2019). Researchers developed nuclease-based PE systems by replacing wild-type SpCas9 with nSpCas9 in order to overcome these limitations. By causing a DSB and thereby activating alternate DNA repair pathways, this method avoids difficult processes involving 5′-flap displacement and heteroduplex resolution. Particularly in genomic areas where nickase-based editors were insufficient, the combination of a 53BP1-inhibitory ubiquitin variation with PEn improved editing effectiveness. However, these approaches also introduced undesired effects, including indels, which exceeded the targeted changes (Li et al., 2023).
The nuclease-based PE systems were enhanced by incorporating paired pegRNA strategies, resulting in the development of Bi-PE with wild-type Cas9 (bi-WT-PE). Nuclease-based Bi-PEs have improved editing efficiencies at genomic locations where nickase-based PE was not successful (Ran et al., 2013). This approach allows for more accurate and precise translocations, inversions, and deletions of long fragments compared with the other gene editing methods. In addition, this system outperforms the performance of wild-type Cas9 systems by effectively avoiding undesired on-target deletions. Nevertheless, these enhancements are often associated with higher rates of insertions and deletions, less accurate modifications at the desired location, and an increased rate of off-target effects, as compared with nick-based PE systems (Jiang et al., 2022).
epegRNA and Bi-PE
The use of epegRNAs in the Bi-PE system has also shown promising outcomes. The only possible exception from this principle could arise if the added length of a 3′ motif may inhibit the synthesis of an excessively long epegRNA or its expression from the U6 promoter. Furthermore, Bi-PE distinguishes itself from previous PE systems without the requirement of the synthesis of nicking sgRNAs. The only feature that requires optimization is a set of epegRNAs, which possess an identical structure to the epegRNAs used for conventional PE. The initial step involves identifying the combinations of protospacer to use. It is important to identify high-efficiency protospacer as predicted by CRISPick. From the list of potential protospacers, it is important to choose pairs of protospacers that are appropriately spaced and located on the opposing strands of DNA. The minimum required distance between the two nicks should be 30 bps in order to avoid steric clashes between the two editor proteins. Research suggests experimenting with around five different protospacer combinations in total. Additionally, the recommended PBS length is 10, 13, or 15 nucleotides for each protospacer. In RTT design, the reverse complement of the desired edit should be on a single epegRNA and the desired modification should be on the other epegRNA. Furthermore, the epegRNA screening for Bi-PE does not use a matrix that combines the lengths of PBS and Reverse transcriptase template (RTT). Instead, it consists of a matrix of epegRNAs for both strands, each of which may have three different potential PBS lengths (Doman, 2023).
One important aspect of experimental design for Bi-PE is that if the intended edit is a deletion, the effectiveness of editing may be excessive due to bias in sample preparation. Although the bias we observed for deletions of 50 bp or less in length was quite moderate, at less than 10%, the bias becomes increasingly obvious as the size of the deletion increases, leading to an overestimation of editing efficiency. Hence, for the purpose of carrying out extensive deletions or ensuring the greatest accuracy in quantification, researchers suggest the utilization of unique molecular identities (UMIs). UMIs, which assign unique barcodes to individual molecules in the initial stage of high-throughput sequencing sample preparation, enable the identification of polymerase chain reaction (PCR) duplicates during downstream processing. Deduplication reduces the bias that occurs during sample preparation and allows for more precise measurement (Doman, 2023).
Therapeutic Potentials of Bi-PE
Scientists conducted a series of experiments to alter genes associated with numerous human disorders with the aim of highlighting the therapeutic potential of Bi-PE. Bi-PE enables the precise rearrangement of chromosomes, which enhances crop development by integrating or arranging beneficial genes. These changes enable the substitution of the introduced gene like Bacillus thuringiensis (Bt) with a novel variant. They also facilitate removal of selective markers and either changing or introducing herbicide tolerant genes in a well-studied plant so as to prevent the development of resistance. Moreover, these modifications allow for simultaneous insertion of multiple favorable genes, hence avoiding the time-consuming process usually required before commercially implementing a new plant type (Awan et al., 2022; Vats et al., 2024).
The Bi-PE method was first validated in mammalian cell culture. A site 3 locus of HEK293T cells was targeted to replace a 90-bp natural sequence with a 38-bp substrate sequence of bacteriophage serine DNA recombinase Bxb1 integrase attB recognition site, facilitating site-specific recombination by an enzyme through the recognition of a precise DNA sequence called the attB recognition site. Researchers used the CleanCap® technology of TriLink to co-transcriptionally cap the pegRNAs. The pegRNAs were engineered to great precision to produce 3′ flaps with overlapping complementarity of length 22–38 bps. A number of pegRNA pairs led to an efficiency of greater than 80% in PE2 RT-mediated transfection of pegRNA pairs. This led to a highly efficient insertion at the attB site (Tao et al., 2022).
Following that, Anzalone and his colleagues used Bi-PE for further investigations. A 108-bp cDNA fragment of FKBP12 was introduced into the HEK3 locus, allowing for the insertion of longer sequences. This cDNA fragment has an insertion efficiency that is 20 times higher than PE3. Interestingly, the Bi-PE was fused to a site-specific integrase for the insertion of larger DNA sequences as well as for easily inserting DNA plasmids greater than 5000 bps into safe-harbor loci. In another experiment, Anzalone and his colleagues validated the integration and inversion of large size DNA, by employing Bi-PE and Bxb1. This recombinase inversion approach allowed for the generation of precise 40 kb sequence inversions. The method used the recombinase inversion technique that produced exogenous 40 kb sequence inversions in high precision. This method might be exploited to treat genetic disorders with loss of function or complex structural defects. The donor plasmids and pegRNAs used in this technique show no genomic homology other than spacer sequences and 8–13 bps to induce reverse transcription. Comprehensive cellular screens may identify DNA repair factors that enhance or diminish the efficiency of Bi-PE editing efficiency, unlocking hidden mechanisms guiding optimization of the method (Anzalone et al., 2022). The Bi-PE method is also used to alter certain target areas of the DMD and PAH genes, which are associated with Duchenne muscular dystrophy (DMD) and phenylketonuria, respectively. The modifications led to a 27% increase in editing efficiency for a 64-bp sequence in phenylketonuria patients and a 23% increase for a 46-bp sequence in DMD patients (Anzalone et al., 2022; Awan et al., 2022; Tao et al., 2022).
The Bi-PE method enhanced the efficiency of simultaneous conversion of multiple bases, but not that of single base conversion compared with PE3. Bi-PE enhanced the efficacy of single base conversions and small fragment deletions or insertions, limited to 2 bps or less (Lin et al., 2021). The recent incorporation of Bi-PE into extensive genomic research has enhanced the functional characterization of genetic variations within their natural setting. Researchers employed the Bi-PE system to execute saturation gene editing (SGE), evaluating and categorizing variations generated by PE. To utilize Bi-PE for SGE, researchers construct a primer library aimed at the specific gene or area and subsequently employ the Bi-PE technique to induce mutations. Their research focused on the NPC intracellular cholesterol transporter-1 gene, mutations of which lead to Niemen–Pick disease type C (Erwood et al., 2022). Chardon et al. developed prime-SGE and evaluated single-nucleotide alterations extensively in oncogenes for their potential to confer drug resistance (Chardon et al., 2023). The identification and classification of large-scale, precise genetic variants by PE will enhance clinical diagnosis, therapy, disease prevention, and genetic counseling, facilitating personalized patient care (Zeng et al., 2024).
Bi-PE has been employed in the treatment of many human genetic disorders, detection of genetic variations, engineering of cellular pathways, and modification of nonhuman species. In addition to its effectiveness in human cells and murine models, PE has been extended to numerous other organisms. Improved PE efficiency was obtained with the use of two pegRNAs in rice and the creation of PlantPegDesigner, a design tool for pegRNA implementation in plants. Zong et al. created plant Plant prime editor (PPE) Engineered plant prime editor (ePPE) by removing the RNase-H domain, including a viral nucleocapsid protein, and employing rational mutagenesis. Bi-PE has been effectively utilized in various organisms, including the moss Physcomitrium patens, sheep, dogs, model vertebrate zebrafish, and key model organism Drosophila melanogaster. These uses highlight the adaptability of PE across several animals (Tao et al., 2022; Zeng et al., 2024).
Bi-PE has also been used in tandem duplication (TD). TDs are frequent structural variations in the genome that have a major role in cancer and genetic diseases. The primary reason for the difficulty in understanding the phenotypic effects caused by TDs is the absence of genetic techniques for modeling these variations. TD by PE (TD-PE) is a novel method that scientists have developed to cause accurate, programmable, and targeted TDs in the mammalian genome. For each TD targeted by the approach, a pair of pegRNAs that encode the identical changes but direct the ssDNA extension in opposite directions must be designed. Reannealing of the modified DNA strands and the replication of the intervening fragment are facilitated by the construction of an RT template for each extension that is homologous to the target area of the alternate sgRNA. It has been shown that the method can produce duplication efficiencies of up to 28.33% for duplications in the range of around 50 bases to 10 kilobases (Jiao et al., 2023; Sun and Gao, 2024).
Potential Limitations and Future Directions of Bi-PE
Bi-PE has expanded genome editing capabilities with the addition of more powerful functions. This review has covered the possibilities of the Bi-PE system along with how these investigations demonstrate its high efficiency and broad variety of editing scopes. Even yet, Bi-PE is not without limitations. When compared with the BE system, the traditional PE system produces less byproducts (Petrova and Smirnikhina, 2023). Bi-PE, like PE, employs pegRNA. The size of the pegRNA continues to be a challenge. It must be compact enough for efficient delivery while containing essential motifs for editing. As researchers strive to enhance Bi-PE editing, the focus on overcoming the size limitation remains of utmost importance. Since pegRNA is usually larger than 100 nucleotides, efficient delivery is challenging. Additionally, its negative charge hinders cellular uptake, and susceptibility to nuclease degradation further complicates delivery. Higher dosages, more off-target effects, decreased editing efficiencies, and restricted therapeutic potentials are the results of ineffective delivery. Researchers use nonviral techniques including lipofection and electroporation, chemical changes to improve pegRNA stability, and viral vectors such as lentiviruses and adeno-associated viruses to address these issues. However, despite these changes, delivery of Bi-PEs remains an issue.
The editing specificity of Bi-PE system varies across a range of variables including temperature, the selection of pegRNA, and the RT template. Editing dual primes also encounters difficulties when handling longer sequences or structural variations. These extensive changes in the genome, such as mutations or insertions that cause diseases, continue to be challenging to correct using this method. Scientists are investigating methods to improve the accuracy of Bi-PE in such cases. The off-target effects of twin PE are significantly reduced in comparison with those of classical CRISPR-Cas9-based approaches. Nonetheless, it is essential to further minimize these effects. Ensuring precise genome editing only at the desired region is important for maintaining safety and accuracy. Improvements in the accuracy and algorithms for predicting pegRNAs design will help reduce the occurrence of undesired effects in nontargeted areas (Anzalone et al., 2022; Awan et al., 2022; Tao et al., 2022).
Researchers strive to overcome the limitations stated above and explore future possibilities in order to fully harness its potential for applications in medicine, agriculture, and other fields. Researchers continue optimizing the PE components including prime editor proteins, pegRNAs and the RT enzyme, MMR pathways, and delivery techniques. Editing rates may be further increased by investigating other RT enzymes or altering the already available ones. It is essential to develop efficient delivery strategies for dual PE tools. Advancements in delivery techniques will provide accurate editing in different kinds of cells and tissues. Nanoparticles, viral vectors, or other carriers may enhance the precise delivery of the editing machinery to specific targets. Optimizing the Bi-PE system from in silico designing to successful delivery into desired cells will improve effectiveness and broaden the scope of editable sequences. Introducing the Bi-PE system into living organisms is an exciting frontier. The use of in vivo applications has the potential to revolutionize gene therapy. This would enable precise modifications of mutations directly inside the cells of patients. The researchers will also prioritize reducing off-target effects while assuring the safety of Bi-PE in real applications. It is believed that Bi-PE will have a significant influence on fundamental research, precision molecular breeding, and disease therapies as this technology continues to be optimized and updated (Awan et al., 2022; Petrova and Smirnikhina, 2023; Vats et al., 2024).
Concluding Remarks
The advancement of CRISPR/Cas-based gene editing continues an evolutionary advancement, with each significant milestone marked by the incorporation of a novel enzymatic mechanism. The current improvements in the Bi-PE-based approach are focused on overcoming the limitation of small-scale editing. Bi-PE offers significant benefits compared with typical CRISPR-Cas9-mediated genome editing in terms of its programmability and flexibility in constructing pegRNAs. Additionally, it minimizes or eliminates off-target effects. Two pegRNAs are used to encode identical edits for both the forward and reverse cases on each DNA strand simultaneously to improve the efficiency of the PE process. Bi-PE may be used for multiplexed genome editing, enabling the improvement of various features in organisms. Bi-PE has the capacity to simultaneously replace and disrupt several targets in the genome by using a combination of pegRNAs. Bi-PE also offers precise integration of large DNA sequences into the genome that can be used for creating transgenic species such as climate-smart crops for ensuring food sustainability and genetically modified animals that possess resistance against certain diseases. Although Bi-PE has several benefits, it still carries a small risk of unwanted indels that cannot be ignored and would not be acceptable for clinical applications. Nevertheless, a well-considered empirical design of the pegRNA may avoid unintended modifications. However, more research and development will be required to improve the editing efficiency and delivery techniques of Bi-PE with the goal to broaden its suitable use in mammals and plants.
Footnotes
Authors’ Contributions
M.S.C. and T.A. conceptualized the article. T.A. and R.M. searched the internet and collected relevant data. T.A. and R.M. wrote the initial draft. M.S.C. and R.M. reviewed the initial version and made required changes. T.A. and R.M. revised the article. T.A. and M.S.C. conceptualized and drafted the figures. All authors approved the final version.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
No funding was received for this article.